Ep 1919867 B1
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-
Compositions for the Reduction of Scarring
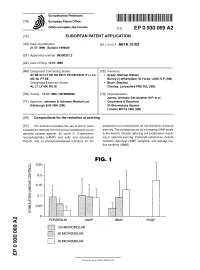
^ ^ ^ ^ I ^ ^ ^ ^ II ^ II ^ ^ ^ ^ ^ ^ ^ ^ ^ ^ ^ ^ ^ ^ ^ I ^ � European Patent Office Office europeen des brevets EP 0 930 069 A2 (12) EUROPEAN PATENT APPLICATION (43) Date of publication: (51) |nt C|.6: A61 K 31/52 21.07.1999 Bulletin 1999/29 (21) Application number: 99300201.3 (22) Date of filing: 12.01.1999 (84) Designated Contracting States: (72) Inventors: AT BE CH CY DE DK ES Fl FR GB GR IE IT LI LU • Grady, Michael William MC NL PT SE Burley-in-Wharfedale, W.Yorks. LS29 7LP (GB) Designated Extension States: • Bloor, Stephen AL LT LV MK RO SI Chorley, Lancashire PR6 7QL (GB) (30) Priority: 13.01.1998 GB 9800640 (74) Representative: James, Anthony Christopher W.P. et al (71) Applicant: Johnson & Johnson Medical Ltd. Carpmaels & Ransford Edinburgh EH2 4NH (GB) 43 Bloomsbury Square London WC1A2RA (GB) (54) Compositions for the reduction of scarring (57) The invention provides the use of one or more preparation of a composition for the reduction of wound substances selected from the group consisting of (a) an scarring. The substances act by increasing cAMP levels adenylyl cyclase agonist, (b) cyclic 3', 5'-adenosine in the wound, thereby reducing cell proliferation result- monophosphate (cAMP) and salts and derivatives ing in reduced scarring. Preferred substances include thereof, and (c) phosphodiesterase inhibitors for the forskolin, dybutyryl cAMP, lisofylline, and isobutyl me- thyl xanthine (IBMX). FIG. 1 0.25-1 o I- z: o 0.2 H o LU > o 0.15H CD < o 0.1 % 0.05 H I 1 I- CM < FORSKOLIN cAMP IBMX PDGF O) CO jjUII 100 MICROMOLAR o 50 MICROMOLAR o CO O) | | 25 MICROMOLAR o a. -

Classification Decisions Taken by the Harmonized System Committee from the 47Th to 60Th Sessions (2011
CLASSIFICATION DECISIONS TAKEN BY THE HARMONIZED SYSTEM COMMITTEE FROM THE 47TH TO 60TH SESSIONS (2011 - 2018) WORLD CUSTOMS ORGANIZATION Rue du Marché 30 B-1210 Brussels Belgium November 2011 Copyright © 2011 World Customs Organization. All rights reserved. Requests and inquiries concerning translation, reproduction and adaptation rights should be addressed to [email protected]. D/2011/0448/25 The following list contains the classification decisions (other than those subject to a reservation) taken by the Harmonized System Committee ( 47th Session – March 2011) on specific products, together with their related Harmonized System code numbers and, in certain cases, the classification rationale. Advice Parties seeking to import or export merchandise covered by a decision are advised to verify the implementation of the decision by the importing or exporting country, as the case may be. HS codes Classification No Product description Classification considered rationale 1. Preparation, in the form of a powder, consisting of 92 % sugar, 6 % 2106.90 GRIs 1 and 6 black currant powder, anticaking agent, citric acid and black currant flavouring, put up for retail sale in 32-gram sachets, intended to be consumed as a beverage after mixing with hot water. 2. Vanutide cridificar (INN List 100). 3002.20 3. Certain INN products. Chapters 28, 29 (See “INN List 101” at the end of this publication.) and 30 4. Certain INN products. Chapters 13, 29 (See “INN List 102” at the end of this publication.) and 30 5. Certain INN products. Chapters 28, 29, (See “INN List 103” at the end of this publication.) 30, 35 and 39 6. Re-classification of INN products. -

Harnessing the Power of P450 Enzymes Aaron T. Larsen
Harnessing the Power of P450 Enzymes A Chemical Auxiliary-Based Approach to Predictable P450 Oxidations at Inactivated C-H Bonds Aaron T. Larsen A thesis submitted to McGill University in partial fulfillment of the requirements of the degree of Doctor of Philosophy Department of Chemistry McGill University Montreal, Quebec, Canada H3A 2K6 Submitted: November, 2011 © Aaron T. Larsen, 2011 Abstract Abstract Enantioselective hydroxylation of one specific methylene in the presence of many similar groups is debatably the most challenging chemical transformation. Although chemists have recently made progress towards the hydroxylation of inactivated C-H bonds, enzymes like P450s (CYPs) remain unsurpassed in specificity and scope. The substrate promiscuity of many P450s is desirable for synthetic applications; however, the inability to predict the products of these enzymatic reactions and the poor activity and stability of these enzymes is impeding advancement. In chapter 2 of this thesis, we evaluate several strategies to improve the activity and stability of CYP3A4. These strategies include the immobilization of CYP3A4 inside molecular hydrogels and silica in addition to the chemical modification of CYP3A4 using various anhydrides. Although none of the strategies we investigate here greatly enhance the catalytic utility of the enzyme, CYP3A4 is shown to be highly tolerant to functionalization at a large number of surface residues and to the presence of extremely high concentrations of silica during catalysis. Recognizing the potential for enzymes containing small, hydrophobic active sites to catalyze Diels-Alder reactions, chapter 3 describes the design and application of several assays to evaluate the Diels-Alderase activity of CYP2E1. Although the presence of CYP2E1 is not found to increase the rates of the reactions we investigate here, the results do demonstrate that there is ii Abstract an interaction between one or more of the substrates and the enzyme at either the active site or at another binding pocket. -

Patent Application Publication ( 10 ) Pub . No . : US 2019 / 0192440 A1
US 20190192440A1 (19 ) United States (12 ) Patent Application Publication ( 10) Pub . No. : US 2019 /0192440 A1 LI (43 ) Pub . Date : Jun . 27 , 2019 ( 54 ) ORAL DRUG DOSAGE FORM COMPRISING Publication Classification DRUG IN THE FORM OF NANOPARTICLES (51 ) Int . CI. A61K 9 / 20 (2006 .01 ) ( 71 ) Applicant: Triastek , Inc. , Nanjing ( CN ) A61K 9 /00 ( 2006 . 01) A61K 31/ 192 ( 2006 .01 ) (72 ) Inventor : Xiaoling LI , Dublin , CA (US ) A61K 9 / 24 ( 2006 .01 ) ( 52 ) U . S . CI. ( 21 ) Appl. No. : 16 /289 ,499 CPC . .. .. A61K 9 /2031 (2013 . 01 ) ; A61K 9 /0065 ( 22 ) Filed : Feb . 28 , 2019 (2013 .01 ) ; A61K 9 / 209 ( 2013 .01 ) ; A61K 9 /2027 ( 2013 .01 ) ; A61K 31/ 192 ( 2013. 01 ) ; Related U . S . Application Data A61K 9 /2072 ( 2013 .01 ) (63 ) Continuation of application No. 16 /028 ,305 , filed on Jul. 5 , 2018 , now Pat . No . 10 , 258 ,575 , which is a (57 ) ABSTRACT continuation of application No . 15 / 173 ,596 , filed on The present disclosure provides a stable solid pharmaceuti Jun . 3 , 2016 . cal dosage form for oral administration . The dosage form (60 ) Provisional application No . 62 /313 ,092 , filed on Mar. includes a substrate that forms at least one compartment and 24 , 2016 , provisional application No . 62 / 296 , 087 , a drug content loaded into the compartment. The dosage filed on Feb . 17 , 2016 , provisional application No . form is so designed that the active pharmaceutical ingredient 62 / 170, 645 , filed on Jun . 3 , 2015 . of the drug content is released in a controlled manner. Patent Application Publication Jun . 27 , 2019 Sheet 1 of 20 US 2019 /0192440 A1 FIG . -

Pentoxifylline As an Adjuvant Treatment in Renal Cell Carcinoma
Pentoxifylline As An Adjuvant Treatment In Renal Cell Carcinoma Item Type text; Electronic Dissertation Authors Mastrandrea, Nicholas Joseph Publisher The University of Arizona. Rights Copyright © is held by the author. Digital access to this material is made possible by the University Libraries, University of Arizona. Further transmission, reproduction or presentation (such as public display or performance) of protected items is prohibited except with permission of the author. Download date 06/10/2021 16:37:11 Link to Item http://hdl.handle.net/10150/337293 PENTOXIFYLLINE AS AN ADJUVANT TREATMENT IN RENAL CELL CARCINOMA by Nicholas J. Mastrandrea A Dissertation Submitted to the Faculty of the DEPARTMENT OF PHARMACOLOGY AND TOXICOLOGY In Partial Fulfillment of the Requirements For the Degree of DOCTOR OF PHILOSOPHY In the Graduate College THE UNIVERSITY OF ARIZONA 2014 1 THE UNIVERSITY OF ARIZONA GRADUATE COLLEGE As members of the Dissertation Committee, we certify that we have read the dissertation prepared by Nicholas J Mastrandrea, titled Pentoxifylline as an Adjuvant Treatment in Renal Cell Carcinoma and recommend that it be accepted as fulfilling the dissertation requirement for the Degree of Doctor of Philosophy. ________________________________________________________ Date: 9/25/14 Serrine S. Lau, Ph. D. ________________________________________________________ Date: 9/25/14 Terrence J. Monks, Ph. D. ________________________________________________________ Date: 9/25/14 Donna Zhang, Ph. D. ________________________________________________________ Date: 9/25/14 Richard Vaillancourt, Ph. D. ________________________________________________________ Date: 9/25/14 George Tsaprailis, Ph. D. Final approval and acceptance of this dissertation is contingent upon the candidate’s submission of the final copies of the dissertation to the Graduate College. I hereby certify that I have read this dissertation prepared under my direction and recommend that it be accepted as fulfilling the dissertation requirement. -

In T Cell Activity. Effects of Selective PDE7 Inhibitors and Dual PDE4/7
International Journal of Molecular Sciences Review Role of Phosphodiesterase 7 (PDE7) in T Cell Activity. Effects of Selective PDE7 Inhibitors and Dual PDE4/7 Inhibitors on T Cell Functions Marianna Szczypka Department of Pharmacology and Toxicology, Faculty of Veterinary Medicine, Wrocław University of Environmental and Life Sciences, Norwida 31, 50-375 Wrocław, Poland; [email protected]; Tel.: +48-71-320-5215 Received: 25 July 2020; Accepted: 22 August 2020; Published: 25 August 2020 Abstract: Phosphodiesterase 7 (PDE7), a cAMP-specific PDE family, insensitive to rolipram, is present in many immune cells, including T lymphocytes. Two genes of PDE7 have been identified: PDE7A and PDE7B with three or four splice variants, respectively. Both PDE7A and PDE7B are expressed in T cells, and the predominant splice variant in these cells is PDE7A1. PDE7 is one of several PDE families that terminates biological functions of cAMP—a major regulating intracellular factor. However, the precise role of PDE7 in T cell activation and function is still ambiguous. Some authors reported its crucial role in T cell activation, while according to other studies PDE7 activity was not pivotal to T cells. Several studies showed that inhibition of PDE7 by its selective or dual PDE4/7 inhibitors suppresses T cell activity, and consequently T-mediated immune response. Taken together, it seems quite likely that simultaneous inhibition of PDE4 and PDE7 by dual PDE4/7 inhibitors or a combination of selective PDE4 and PDE7 remains the most interesting therapeutic target for the treatment of some immune-related disorders, such as autoimmune diseases, or selected respiratory diseases. -

Lista De Productos En Orden Alfabético
FARMACÉUTICO QUÍMICO VETERINARIO COSMÉTICO Lista de productos en orden alfabético ALUMINIUM SULPHATE A AMBROXOL HCL AMBROXOL HCL PELLETS 25% AMIFOSTINE ABACAVIR AMILORIDE HCL ABACAVIR SULPHATE AMINACRINE HYDROCHLORIDE ACEBROPHYLLINE AMINOPHYLLINE ACEBUTOLOL HCL AMITRIPTYLINE EMBONATE ACECLOFENAC AMITRIPTYLINE HCL ACEFYLLINE AMITRIPTYLINE N OXIDE ACEFYLLINE PIPERAZINE ORAL AMLA EXTRACT ACEFYLLINE PIPERAZINE - STERILE) AMLODIPINE BESYLATE ACENOCOUMAROL (NICOUMALONE) AMLODIPINE MALEATE ACEPHATE TECHNICAL AMLODIPINE MESYLATE ACEPHYLLINE PIPERAZINE AMMONIUM ACEPIPHYLLINE / ACEPIFYLLINE AMMONIUM ACETATE ACETAMINOPHEN/PARACETAMOL AMMONIUM CHLORIDE ACID YELLOW 73 AMMONIUM DICHROMATE ACITRETIN AMMONIUM HYPOPHOSPHATE ACRIFLAVINE HYDROCHLORIDE AMMONIUM IODIDE ACRIFLAVINE NEUTRAL AMMONIUM PHOSPHATE DIBASIC ACRINOL AMMONIUM PHOSPHATE MONOBASIC ALUMINIUM OXIDE AMMONIUM SULPHATE ACTIVATED CHARCOL AMOMUM SUBULATUM ACYCLOVIR AMOXAPINE ACYCLOVIR SODIUM AMOXYCILLIN TRIHYDRATE ADAPALENE AMPICILLIN ANHYDROUS ADEFOVIR DIPIVOXIL AMPICILLIN SODIUM AND SULBACTAM SODIUM ADHATODA VASICAL LEAF AMPICILLIN SODIUM+SULBACTAMSODIUM AEGLE MARMELOUS FRUIT AMPICILLIN TRIHYDRATE AGOMELATINE AMPIROXICAM AJOWAN SEED OLEORESIN AMPRENAVIR AJOWAN OIL ANACYCLUS PYRETHRUM ALBENDAZOLE ANAGRELIDE HCL ALBUTEROL SULPHATE ANAGRELIDE HCL MONOHYDRATE GAMMA UNDECALACTONE ANASTRAZOLE ALENDRONATE SODIUM ANDROGRAPHATIS ALFUZOSIN ANDROGRAPHIS PANICULATA ALFUZOSIN HCL ANDROGRAPHOLIDE ALLIUM CEPA BULBS ANESTHETIC ETHER ALLIUM SATIVUM BULBS ANILINE HYDROCHLORIDE ALLYL HEPTANOATE ANISE OIL ALMAGATE -

Stembook 2018.Pdf
The use of stems in the selection of International Nonproprietary Names (INN) for pharmaceutical substances FORMER DOCUMENT NUMBER: WHO/PHARM S/NOM 15 WHO/EMP/RHT/TSN/2018.1 © World Health Organization 2018 Some rights reserved. This work is available under the Creative Commons Attribution-NonCommercial-ShareAlike 3.0 IGO licence (CC BY-NC-SA 3.0 IGO; https://creativecommons.org/licenses/by-nc-sa/3.0/igo). Under the terms of this licence, you may copy, redistribute and adapt the work for non-commercial purposes, provided the work is appropriately cited, as indicated below. In any use of this work, there should be no suggestion that WHO endorses any specific organization, products or services. The use of the WHO logo is not permitted. If you adapt the work, then you must license your work under the same or equivalent Creative Commons licence. If you create a translation of this work, you should add the following disclaimer along with the suggested citation: “This translation was not created by the World Health Organization (WHO). WHO is not responsible for the content or accuracy of this translation. The original English edition shall be the binding and authentic edition”. Any mediation relating to disputes arising under the licence shall be conducted in accordance with the mediation rules of the World Intellectual Property Organization. Suggested citation. The use of stems in the selection of International Nonproprietary Names (INN) for pharmaceutical substances. Geneva: World Health Organization; 2018 (WHO/EMP/RHT/TSN/2018.1). Licence: CC BY-NC-SA 3.0 IGO. Cataloguing-in-Publication (CIP) data. -

A Abacavir Abacavirum Abakaviiri Abagovomab Abagovomabum
A abacavir abacavirum abakaviiri abagovomab abagovomabum abagovomabi abamectin abamectinum abamektiini abametapir abametapirum abametapiiri abanoquil abanoquilum abanokiili abaperidone abaperidonum abaperidoni abarelix abarelixum abareliksi abatacept abataceptum abatasepti abciximab abciximabum absiksimabi abecarnil abecarnilum abekarniili abediterol abediterolum abediteroli abetimus abetimusum abetimuusi abexinostat abexinostatum abeksinostaatti abicipar pegol abiciparum pegolum abisipaaripegoli abiraterone abirateronum abirateroni abitesartan abitesartanum abitesartaani ablukast ablukastum ablukasti abrilumab abrilumabum abrilumabi abrineurin abrineurinum abrineuriini abunidazol abunidazolum abunidatsoli acadesine acadesinum akadesiini acamprosate acamprosatum akamprosaatti acarbose acarbosum akarboosi acebrochol acebrocholum asebrokoli aceburic acid acidum aceburicum asebuurihappo acebutolol acebutololum asebutololi acecainide acecainidum asekainidi acecarbromal acecarbromalum asekarbromaali aceclidine aceclidinum aseklidiini aceclofenac aceclofenacum aseklofenaakki acedapsone acedapsonum asedapsoni acediasulfone sodium acediasulfonum natricum asediasulfoninatrium acefluranol acefluranolum asefluranoli acefurtiamine acefurtiaminum asefurtiamiini acefylline clofibrol acefyllinum clofibrolum asefylliiniklofibroli acefylline piperazine acefyllinum piperazinum asefylliinipiperatsiini aceglatone aceglatonum aseglatoni aceglutamide aceglutamidum aseglutamidi acemannan acemannanum asemannaani acemetacin acemetacinum asemetasiini aceneuramic -

31St International Symposium on Intensive Care and Emergency Medicine
Critical Care 2011, Volume 15 Suppl 1 http://ccforum.com/supplements/15/S1 MEETING ABSTRACTS 31st International Symposium on Intensive Care and Emergency Medicine Brussels, Belgium, 22-25 March 2011 Published: 1 March 2011 P1 QT dispersion has been suggested to give information about the Eff ects of thyroid hormones on major cardiovascular risk in acute heterogeneity of myocardial repolarization. coronary syndromes Methods Our study included 60 patients presented with acute A Bayrak1, A Bayır2, K Uçar Karabulut3 STEMI, the study populations were divided into two groups: Group 1Selçuk University, Meram Faculty of Medicine, Konya, Turkey; 2Selçuk I: 30 patients who underwent primary PCI. Group II: 15 patients who University, Selçuklu Faculty of Medicine, Emergency Department, Konya, received streptokinase. Group III: 15 patients who did not receive Turkey; 3Emergency Sercice of Şırnak State Hospital, Şırnak, Turkey reperfusion therapy. QTd and QTcd were measured and compared in Critical Care 2011, 15(Suppl 1):P1 (doi: 10.1186/cc9421) the three groups on admission, after 24 hours and after 5 days. Results QTd and QTcd were signifi cantly higher in patients with anterior Introduction In this study we aimed to investigate the relationship compared with inferior MI (79.16 ± 25.67 ms vs. 62 ± 18.17 ms, P = 0.004 between thyroid hormone abnormalities and major cardiovascular regarding QTd and 91.95 ± 28.76 ms vs. 68.33 ± 23.52 ms, P <0.001 events and sudden cardiac death at 3 and 6 months after discharge in regarding QTcd). After 24 hours, QTd and QTcd were signifi cantly lower patients who were admitted to the Emergency Department with acute in group I than groups II and III (34.33 ± 13.56 ms vs. -

Phosphodiesterase Inhibitors in Acute Lung Injury: What Are the Perspectives?
International Journal of Molecular Sciences Review Phosphodiesterase Inhibitors in Acute Lung Injury: What Are the Perspectives? Daniela Mokra 1,* and Juraj Mokry 2 1 Department of Physiology, Jessenius Faculty of Medicine in Martin, Comenius University in Bratislava, 03601 Martin, Slovakia 2 Department of Pharmacology, Jessenius Faculty of Medicine in Martin, Comenius University in Bratislava, 03601 Martin, Slovakia; [email protected] * Correspondence: [email protected] Abstract: Despite progress in understanding the pathophysiology of acute lung damage, currently approved treatment possibilities are limited to lung-protective ventilation, prone positioning, and supportive interventions. Various pharmacological approaches have also been tested, with neuro- muscular blockers and corticosteroids considered as the most promising. However, inhibitors of phosphodiesterases (PDEs) also exert a broad spectrum of favorable effects potentially beneficial in acute lung damage. This article reviews pharmacological action and therapeutical potential of nonselective and selective PDE inhibitors and summarizes the results from available studies focused on the use of PDE inhibitors in animal models and clinical studies, including their adverse effects. The data suggest that xanthines as representatives of nonselective PDE inhibitors may reduce acute lung damage, and decrease mortality and length of hospital stay. Various (selective) PDE3, PDE4, and PDE5 inhibitors have also demonstrated stabilization of the pulmonary epithelial–endothelial barrier -

(12) United States Patent (10) Patent No.: US 9,181.233 B2 Heiser Et Al
US009181233B2 (12) United States Patent (10) Patent No.: US 9,181.233 B2 Heiser et al. (45) Date of Patent: Nov. 10, 2015 2007/0191366 A1* 8, 2007 Hoffmann et al. ......... 514,235.5 (54) INHIBITORS OF GLUTAMINYLCYCLASE 2008/0214620 A1* 9, 2008 Heiser et al. ....... ... 514,338 (75) Inventors: Ulrich Heiser, Halle/Saale (DE); Daniel 2010.0040575 A1 2/2010 Hoffmann et al. ........... 424/85.2 Ramsbeck, Halle/Saale (DE); Torsten 2010 OO69381 A1 3, 2010 Itoh Hoffmann, Halle/Saale (DE); Livia FOREIGN PATENT DOCUMENTS Boehme, Halle/Saale (DE); Hans-Ulrich Demuth, Halle/Saale (DE) EP 1375486 1, 2004 WO WO 2004/005.283 1, 2004 (73) Assignee: PROBIODRUGAG. Halle/Saale (DE) WO WO 2004/O14881 2, 2004 WO WO 2004/031177 4/2004 WO WO2005/121132 12/2005 (*) Notice: Subject to any disclaimer, the term of this WO WO 2007/OO3961 1, 2007 patent is extended or adjusted under 35 WO WO 2008/O16123 2, 2008 U.S.C. 154(b) by 224 days. WO WO 2008/042571 4/2008 WO WO2008/065141 6, 2008 (21) Appl. No.: 13/040,159 WO WO 2008.154241 12/2008 WO WO 2008,157 273 12/2008 WO WO 2009/051705 4/2009 (22) Filed: Mar. 3, 2011 WO WO 2009, 111442 9, 2009 (65) Prior Publication Data OTHER PUBLICATIONS US 2011 FO224259 A1 Sep. 15, 2011 International Application Published as WO2005/121132 on Jun. 5, 2008, abstract in English only. Related U.S. Application Data Hess et al., 1-(5-Carboxy- and 5-carbamoylindol-lyl)propane-2- ones as inhibitors of human cytosolic phospholipase A2a: (60) Provisional application No.