Model of Gene Expression in Extreme Cold
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Reference Transcriptome for the High- Antarctic Cryopelagic Notothenioid Fish Pagothenia Borchgrevinki Kevin T Bilyk1,2* and C-H Christina Cheng1
Bilyk and Cheng BMC Genomics 2013, 14:634 http://www.biomedcentral.com/1471-2164/14/634 RESEARCH ARTICLE Open Access Model of gene expression in extreme cold - reference transcriptome for the high- Antarctic cryopelagic notothenioid fish Pagothenia borchgrevinki Kevin T Bilyk1,2* and C-H Christina Cheng1 Abstract Background: Among the cold-adapted Antarctic notothenioid fishes, the high-latitude bald notothen Pagothenia borchgrevinki is particularly notable as the sole cryopelagic species, exploiting the coldest and iciest waters of the Southern Ocean. Because P. borchgrevinki is a frequent model for investigating notothenioid cold-adaptation and specialization, it is imperative that “omic” tools be developed for this species. In the absence of a sequenced genome, a well annotated reference transcriptome of the bald notothen will serve as a model of gene expression in the coldest and harshest of all polar marine environments, useful for future comparative studies of cold adaptation and thermal responses in polar teleosts and ectotherms. Results: We sequenced and annotated a reference transcriptome for P. borchgrevinki, with added attention to capturing the transcriptional responses to acute and chronic heat exposures. We sequenced by Roche 454 a normalized cDNA library constructed from pooled mRNA encompassing multiple tissues taken from environmental, warm acclimating, and acute heat stressed specimens. The resulting reads were assembled into 42,620 contigs, 17,951 of which could be annotated. We utilized this annotated portion of the reference transcriptome to map short Illumina reads sequenced from the gill and liver of environmental specimens, and also compared the gene expression profiles of these two tissue transcriptomes with those from the temperate model fish Danio rerio. -

Induction of Heat Shock Proteins in Cold- Adapted and Cold
CORE Metadata, citation and similar papers at core.ac.uk Provided by ScholarWorks@UA INDUCTION OF HEAT SHOCK PROTEINS IN COLD- ADAPTED AND COLD- ACCLIMATED FISHES By Laura Elizabeth Teigen Dr. Kristin O'Brien Advisory Committee Chair Dr. Diane Wagner Chair, Department of Biology and Wildlife APPROVED: ;t.-/ INDUCTION OF HEAT SHOCK PROTEINS IN COLD- ADAPTED AND COLD- ACCLIMATED FISHES A THESIS Presented to the Faculty of the University of Alaska Fairbanks in Partial Fulfillment of the Requirements for the Degree of MASTER OF SCIENCE By Laura Elizabeth Teigen, B.A. Fairbanks, Alaska May 2014 v Abstract I examined the effects of oxidative stress and changes in temperature on heat shock protein (Hsp) levels in cold-adapted and cold-acclimated fishes. Adaptation of Antarctic notothenioids to cold temperature is correlated with high levels of Hsps, thought to minimize cold-induced protein denaturation. Hsp70 levels were measured in red- and white-blooded Antarctic notothenioid fishes exposed to their critical thermal maximum (CTMax), 4C warm acclimated, and notothenioids from different latitudes. I determined the effect of cold acclimation on Hsp levels and the role of sirtuins in regulating Hsp expression and changes in metabolism in threespine stickleback, Gasterosteus aculeatus, cold-acclimated to 8C. Levels of Hsps do not increase in Antarctic notothenioids exposed to their CTMax, and warm acclimation reduced levels of Hsp70. Hsp70 levels were higher in Antarctic notothenioids compared to a temperate notothenioid and higher in white-blooded notothenioids compared to red-blooded notothenioids, despite higher oxidative stress levels in red-blooded fish, suggesting Hsp70 does not mitigate oxidative stress. -

Evaluation of Thermal Stress in Tropical Marine Organisms in the Context of Climate Warming
UNIVERSIDADE DE LISBOA FACULDADE DE CIÊNCIAS Evaluation of thermal stress in tropical marine organisms in the context of climate warming Doutoramento em Biologia Especialidade em Biologia Marinha e Aquacultura Sara Carolina Gusmão Coito Madeira Tese orientada por: Doutora Catarina Vinagre Professor Doutor Mário Diniz Professor Doutor Henrique Cabral Documento especialmente elaborado para a obtenção do grau de doutor 2018 2018 UNIVERSIDADE DE LISBOA FACULDADE DE CIÊNCIAS Evaluation of thermal stress in tropical marine organisms in the context of climate warming Doutoramento em Biologia Especialidade em Biologia Marinha e Aquacultura Sara Carolina Gusmão Coito Madeira Tese orientada por: Doutora Catarina Vinagre Professor Doutor Mário Diniz Professor Doutor Henrique Cabral Documento especialmente elaborado para a obtenção do grau de doutor 2018 2018 UNIVERSIDADE DE LISBOA FACULDADE DE CIÊNCIAS Evaluation of thermal stress in tropical marine organisms in the context of climate warming Doutoramento em Biologia Especialidade em Biologia Marinha e Aquacultura Sara Carolina Gusmão Coito Madeira Tese orientada por: Doutora Catarina Vinagre Professor Doutor Mário Diniz Professor Doutor Henrique Cabral Júri: Presidente: ● Doutora Maria Manuela Gomes Coelho de Noronha Trancoso, Professora Catedrática e Membro do Conselho Científico, Faculdade de Ciências da Universidade de Lisboa Vogais: ● Doutor Luís Miguel dos Santos Russo Vieira, Investigador Auxiliar, Centro Interdisciplinar de Investigação Marinha e Ambiental (CIIMAR) da Universidade do Porto -

THREE PINNIPED SPECIES, by Susan D. Inglis, MS a Dissertation
Dietary effects on protein turnover in three pinniped species, Eumetopias jubatus, Phoca vitulina, and Leptonychotes weddellii Item Type Thesis Authors Inglis, Susan D. Download date 11/10/2021 13:32:47 Link to Item http://hdl.handle.net/11122/10505 DIETARY EFFECTS ON PROTEIN TURNOVER IN THREE PINNIPED SPECIES, EUMETOPIAS JUBATUS, PHOCA VITULINA, AND LEPTONYCHOTES WEDDELLII By Susan D. Inglis, MS A Dissertation Submitted in Partial Fulfillment of the Requirements for the Degree of Doctor of Philosophy in Marine Biology University of Alaska Fairbanks May 2019 ©2019 Susan D. Inglis APPROVED: Michael Castellini, Committee Chair Shannon Atkinson, Committee Member Perry Barboza, Committee Member James Carpenter, Committee Member Lorrie Rea, Committee Member Matthew Wooller, Chair Department of Marine Biology Bradley S. Moran, Dean College of Fisheries and Ocean Sciences Michael Castellini, Dean of the Graduate School Abstract The role of dietary protein in pinniped (seal and sea lion) nutrition is poorly understood. Although these marine mammals derive the majority of their daily energetic needs from lipid, lipids cannot supply essential amino acids which have to come from protein fractions of the diet. Protein regulation is vital for cellular maintenance, molt, fasting metabolism, exercise and development. Proteins are composed of linked amino acids (AA), and net protein turnover is the balance between protein synthesis from component AA, and degradation back to AA. Protein regulation is influenced by dietary intake and quality, as well as physiological and metabolic requirements. In this work, pinniped diet quality was assessed through comparisons of amino acid profiles between maternal milk, blood serum, and seasonal prey of wild juvenile Steller sea lions (Eumetopias jubatus) in Southcentral Alaska. -

The Importance of Antarctic Toothfish As Prey of Weddell Seals in the Ross
Antarctic Science 21(4), 317–327 (2009) & Antarctic Science Ltd 2009 doi:10.1017/S0954102009001953 Review The importance of Antarctic toothfish as prey of Weddell seals in the Ross Sea DAVID G. AINLEY1* and DONALD B. SINIFF2 1HT Harvey & Associates, Los Gatos, CA 95032, USA 2Department of Ecology, Evolution and Behavioral Biology, University of Minnesota, St Paul, MN 55108, USA *[email protected] Abstract: Uncertainty exists over the importance of Antarctic toothfish (Dissostichus mawsoni) as prey of top predators in the Ross Sea. In this paper we assess relative weight given to direct, observational evidence of prey taken, as opposed to indirect evidence from scat and biochemical analysis, and conclude that toothfish are important to Weddell seals (Leptonychotes weddellii). The seals eat only the flesh of large toothfish and therefore they are not detected in scat or stomach samples; biochemical samples have been taken from seal sub-populations where toothfish seldom occur. Using direct observations of non-breeding seals away from breeding haulouts in McMurdo Sound, 0.8–1.3 toothfish were taken per day. Based on these and other data, the non-breeding portion of the McMurdo Sound seal population, during spring and summer, consume about 52 tonnes of toothfish. Too many unknowns exist to estimate the non-trivial amount consumed by breeders. We discuss why reduced toothfish availability to Weddell seals, for energetic reasons, cannot be compensated by a switch to silverfish (Pleuragramma antarcticum) or squid. The Ross Sea toothfish fishery should be reduced including greater spatial management, with monitoring of Weddell seal populations by CCAMLR. Otherwise, probable cascades will lead to dramatic changes in the populations of charismatic megafauna. -
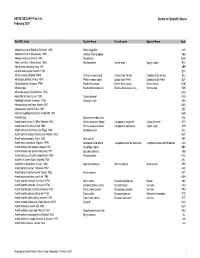
ASFIS ISSCAAP Fish List February 2007 Sorted on Scientific Name
ASFIS ISSCAAP Fish List Sorted on Scientific Name February 2007 Scientific name English Name French name Spanish Name Code Abalistes stellaris (Bloch & Schneider 1801) Starry triggerfish AJS Abbottina rivularis (Basilewsky 1855) Chinese false gudgeon ABB Ablabys binotatus (Peters 1855) Redskinfish ABW Ablennes hians (Valenciennes 1846) Flat needlefish Orphie plate Agujón sable BAF Aborichthys elongatus Hora 1921 ABE Abralia andamanika Goodrich 1898 BLK Abralia veranyi (Rüppell 1844) Verany's enope squid Encornet de Verany Enoploluria de Verany BLJ Abraliopsis pfefferi (Verany 1837) Pfeffer's enope squid Encornet de Pfeffer Enoploluria de Pfeffer BJF Abramis brama (Linnaeus 1758) Freshwater bream Brème d'eau douce Brema común FBM Abramis spp Freshwater breams nei Brèmes d'eau douce nca Bremas nep FBR Abramites eques (Steindachner 1878) ABQ Abudefduf luridus (Cuvier 1830) Canary damsel AUU Abudefduf saxatilis (Linnaeus 1758) Sergeant-major ABU Abyssobrotula galatheae Nielsen 1977 OAG Abyssocottus elochini Taliev 1955 AEZ Abythites lepidogenys (Smith & Radcliffe 1913) AHD Acanella spp Branched bamboo coral KQL Acanthacaris caeca (A. Milne Edwards 1881) Atlantic deep-sea lobster Langoustine arganelle Cigala de fondo NTK Acanthacaris tenuimana Bate 1888 Prickly deep-sea lobster Langoustine spinuleuse Cigala raspa NHI Acanthalburnus microlepis (De Filippi 1861) Blackbrow bleak AHL Acanthaphritis barbata (Okamura & Kishida 1963) NHT Acantharchus pomotis (Baird 1855) Mud sunfish AKP Acanthaxius caespitosa (Squires 1979) Deepwater mud lobster Langouste -

Gymnelus Hemifasciatus
Overview of polar exhibit tanks and List of the creatures in house Keisuke ICHIKAWA, Takashi KIFUNE, Ryousuke KOMI, Mayuko MURAMATSU Shota HABA, Mayuka ISHIGAMI, Kokoro SATO, Keita SEKI Tokyo Sea Life Park ● Exhibition area of “Arctic and Antarctic Oceans” The first Antarctic Ocean tank “Clumps The Tokyo Sea Life Park has been exhibiting polar of submerged drifting algae” creatures since the aquarium opened in 1989. In the Antarctic Ocean, it takes a long time These exhibits have featured creatures from the for broken up seaweed to decompose Antarctic Ocean since 1989, and creatures from the because the seawater is so cold. The clumps Arctic Ocean since 1992. Exhibits include panels of submerged drifting algae are home to all introducing the differences between the Arctic and sorts of creatures. For example, Glyptonotus Antarctic environments and their respective antarcticus and small fish of Nototheniidae creatures, as well as showing how these creatures use those as a place to hide or find food. were collected, and the formalin specimen of Fig. 4 “Antarctic Ocean 1” Tank Dissostichus mawsoni. There are three tanks. Two The second Antarctic Ocean tank of these tanks measure 112 cm × 95 cm × 89 cm “Nototheniidae” (volume: 0.8 m3). The other tank measures 157 cm Many creatures living in polar regions have Fig. 1 Exhibition area × 95 cm × 89 cm (volume: 0.7 m3). The latter tank antifreeze proteins in their body. This can be divided into three parts due to the includes Nototheniidae, a significant fish exhibition of small creatures. family in Antarctic regions. Nototheniidae possess antifreeze glycopeptide to prevent the formation of ice crystals in their bodily fluids. -

Land Use Drives the Physiological Properties of a Stream Fish
LAND USE DRIVES THE PHYSIOLOGICAL PROPERTIES OF A STREAM FISH BY ZACHARY WILLIAM BLEVINS THESIS Submitted in partial fulfillment of the requirements for the degree of Master of Science in Natural Resources and Environmental Sciences in the Graduate College of the University of Illinois at Urbana-Champaign, 2012 Urbana, Illinois Advisers: Assistant Professor Cory D. Suski, Director of Research Research Professor David H. Wahl ii ABSTRACT Human activities within the riparian zone can alter the abiotic properties of watersheds and stream reaches, potentially resulting in abiotic conditions that are stressful for resident fishes. The inability of fish to cope physiologically with stressful abiotic conditions can have deleterious effects on individuals, and could potentially lead to population declines or changes to community structure. Defining links between the landscape and performance of individual stream fishes can therefore improve our ability to predict how land use changes can impact stream communities, which has relevance for management activities. These studies tested the hypothesis that land cover at the watershed and reach scale influence the physiological stress response of a resident stream fish. For this, replicate streams in agricultural watersheds and forested watersheds were identified for the watershed-scale study and replicate forested and agricultural stream reaches were identified for the reach-scale study. In both the watershed and reach-scale studies, sampling demonstrated that streams in agricultural watersheds and stream reaches were warmer and more thermally variable than those from forested areas. In the watershed-scale study, creek chub from each land use type were sampled for blood and muscle in the field, following exposure to thermal and oxygen stressors in the laboratory, and after prolonged holding at elevated temperatures that replicated field conditions. -

SOUTHERN OCEAN Card Games
SOUTHERN OCEAN card games The Southern Ocean is the ocean around Antarctica. It extends out far enough to take in several groups of “sub-antarctic islands such as South Georgia Island, the South Sandwich Islands, Kerguelen Islands, and the South Orkney Islands. This game focuses as much as possible on the species that live on or very near to the continent of Antarctica itself. For examples, the penguins listed here are the only species that live on the continent year round. VERY IMPORTANT NOTES: 1) These cards do not represent a complete survey of all animals found in Antarctic waters. For ex- ample, salps, urchins and clams do not appear in the game. For a complete fi eld guide to what lives in and around the Ross Ice Shelf, try this website: http://www.peterbrueggeman.com/nsf/fguide/index.html Another very good site is: http://www.antarctica.gov.au/about-antarctica/wildlife/animals 2) Please don’t use these cards as the last word on the Antarctic food web. This is especially true for the animals that eat a lot of things. If they are not picky eaters, it’s hard to list everything they will eat. (This is especially true for anemones, worms, jellies and sea spiders.) Also, some of these animals have subspecies that differ in what they eat. This is especially true for the fi sh, sea cucumbers, sea stars, and crustaceans. Even within a species you might fi nd some in one area eating a slightly different diet than those in another area. Also, diets can vary at different times of the year or during years when a particular type of food is scarce. -

Phylogenetic Analysis of Antarctic Notothenioids Illuminates the Utility of 7 Radseq for Resolving Cenozoic Adaptive Radiations
0ROHFXODU3K\ORJHQHWLFVDQG(YROXWLRQ ² Contents lists available at ScienceDirect Molecular Phylogenetics and Evolution journal homepage: www.elsevier.com/locate/ympev Phylogenetic analysis of Antarctic notothenioids illuminates the utility of 7 RADseq for resolving Cenozoic adaptive radiations Thomas J. Neara,b,⁎, Daniel J. MacGuigana, Elyse Parkera, Carl D. Struthersc, Christopher D. Jonesd, Alex Dornburge a Department of Ecology & Evolutionary Biology, Yale University, P.O. Box 208106, New Haven, CT 06520, USA b Peabody Museum of Natural History, Yale University, New Haven, CT 06520, USA c Museum of New Zealand Te Papa Tongarewa, Wellington, New Zealand d Antarctic Ecosystem Research Division, NOAA Southwest Fisheries Science Center, La Jolla, CA 92037, USA e North Carolina Museum of Natural Sciences, Raleigh, NC 27601, USA ARTICLE INFO ABSTRACT Keywords: Notothenioids are a clade of ∼120 species of marine fishes distributed in extreme southern hemisphere tem- Notothenioidei perate near-shore habitats and in the Southern Ocean surrounding Antarctica. Over the past 25 years, molecular Phylogenomics and morphological approaches have redefined hypotheses of relationships among notothenioid lineages as well Phylogenetic informativeness as their relationships among major lineages of percomorph teleosts. These phylogenies provide a basis for in- Signal noise vestigation of mechanisms of evolutionary diversification within the clade and have enhanced our understanding Icefishes of the notothenioid adaptive radiation. Despite extensive efforts, there remain several questions concerning the phylogeny of notothenioids. In this study, we deploy DNA sequences of ∼100,000 loci obtained using RADseq to investigate the phylogenetic relationships of notothenioids and to assess the utility of RADseq loci for lineages that exhibit divergence times ranging from the Paleogene to the Quaternary. -

Argentina–Chile National Geographic Pristine Seas Expedition to the Antarctic Peninsula
ARGENTINA–CHILE NATIONAL GEOGRAPHIC PRISTINE SEAS EXPEDITION TO THE ANTARCTIC PENINSULA SCIENTIFIC REPORT 2019 1 Pristine Seas, National Geographic Society, Washington, DC, USA 2 Hawaii Institute of Marine Biology, University of Hawaii, Kaneohe, Hawaii, USA 3 Charles Darwin Research Station, Charles Darwin Foundation, Puerto Ayora, Galápagos, Ecuador 4 Centre d’Estudis Avancats de Blanes-CSIC, Blanes, Girona, Spain 5 Exploration Technology, National Geographic Society, Washington, DC, USA 6 Instituto Antártico Argentino/Dirección Nacional del Antártico, Cancilleria Argentina, Buenos Aires, Argentina. 7 Departamento Científico, Instituto Antártico Chileno, Punta Arenas, Chile 8 Fundación Ictiológica, Santiago, Chile 9 Instituto de Diversidad y Ecología Animal (IDEA), CONICET- UNC and Facultad de Ciencias Exactas, Físicas y Naturales, Universidad Nacional de Córdoba, Córdoba, Argentina 10 Laboratorio de Ictioplancton (LABITI), Escuela de Biología Marina, Facultad de Ciencias del Mar y de Recursos Naturales, Universidad de Valparaíso, Viña del Mar, Chile 11 The Pew Charitable Trusts & Antarctic and Southern Ocean Coalition, Washington DC CITATION: Friedlander AM1,2, Salinas de León P1,3, Ballesteros E4, Berkenpas E5, Capurro AP6, Cardenas CA7, Hüne M8, Lagger C9, Landaeta MF10, Santos MM6, Werner R11, Muñoz A1. 2019. Argentina– Chile–National Geographic Pristine Seas Expedition to the Antarctic Peninsula. Report to the governments of Argentina and Chile. National Geographic Pristine Seas, Washington, DC 84pp. TABLE OF CONTENTS EXECUTIVE SUMMARY . 3 INTRODUCTION . 9 1 .1 . Geology of the Antarctic Peninsula 1 .2 . Oceanography (Antarctic Circumpolar Current) 1 .3 . Marine Ecology 1 .4 . Antarctic Governance 1 .5 . Current Research by Chile and Argentina EXPLORING THE BIODIVERSITY OF THE ANTARCTIC PENINSULA: ONE OF THE LAST OCEAN WILDERNESSES . -

Persistent Organic Pollutants in Benthic and Pelagic Organisms Off Adélie Land, Antarctica ⇑ A
Marine Pollution Bulletin 77 (2013) 82–89 Contents lists available at ScienceDirect Marine Pollution Bulletin journal homepage: www.elsevier.com/locate/marpolbul Persistent organic pollutants in benthic and pelagic organisms off Adélie Land, Antarctica ⇑ A. Goutte a,c, , M. Chevreuil b, F. Alliot b, O. Chastel c, Y. Cherel c, M. Eléaume d, G. Massé a,e a LOCEAN/IPSL, UMR7159, Université Pierre et Marie Curie, 75005 Paris, France b Laboratoire Hydrologie Environnement, Ecole Pratique Des Hautes Etudes, UMR Sisyphe, Université Pierre et Marie Curie, 75005 Paris, France c Centre d’Etudes Biologiques de Chizé, CNRS UPR 1934, 79360 Villiers-en-Bois, France d Muséum National d’Histoire Naturelle, Département Milieux et Peuplements Aquatiques, UMR 7208 BOREA, CP26, 57 Rue Cuvier, 75231 Paris Cedex 05, France e TAKUVIK, UMI3376 CNRS, Université Laval, G1V0A6 Québec, Canada article info abstract Keywords: The concentrations of polychlorinated biphenyls (PCB), hexachlorobenzene (HCB), pentachlorobenzene Polar ecosystem (PeCB) and polybrominated diphenylethers (PBDE) were described in benthic and pelagic species col- Persistent organic pollutants lected off Adélie Land, Antarctica. Strong differences were observed among species, with reduced PeCB Echinoderm and HCB levels in benthic species, and elevated PCB levels in the Antarctic yellowbelly rockcod, the Fish Antarctic sea urchin and the snow petrel. Lower-chlorinated congeners were predominant in krill; Krill penta-PCBs in benthic organisms; hexa- and hepta-PCBs in seabirds and cryopelagic fish. This segregation Seabird may result from sedimentation process, specific accumulation and excretion, and/or biotransformation processes. The presence of PBDEs in Antarctic coastal organisms may originate from atmospheric trans- port and partly from a contamination by local sources.