Heterogeneity in Combined Immunodeficiencies with Associated Or Syndromic Features (Review)
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Exome Sequencing Identifies Mutations in the Gene TTC7A In
Developmental defects J Med Genet: first published as 10.1136/jmedgenet-2012-101483 on 19 February 2013. Downloaded from ORIGINAL ARTICLE Exome sequencing identifies mutations in the gene TTC7A in French-Canadian cases with hereditary multiple intestinal atresia Mark E Samuels,1 Jacek Majewski,2 Najmeh Alirezaie,2 Isabel Fernandez,1,3 Ferran Casals,1 Natalie Patey,1,4 Hélène Decaluwe,1,5 Isabelle Gosselin,6 Elie Haddad,1,3,5 Alan Hodgkinson,1 Youssef Idaghdour,1 Valerie Marchand,1,5 1,5 6,7 6 6 Open Access Jacques L Michaud, Marc-André Rodrigue, Sylvie Desjardins, Stéphane Dubois, Scan to access more 1,3 1,5 6,7 8 free content Francoise Le Deist, Philip Awadalla, Vincent Raymond, Bruno Maranda ▸ Additional material is ABSTRACT attempted, outcomes are poor and the condition is published online only. To view Background Congenital multiple intestinal atresia usual fatal within the first month of life. To date, please visit the journal online (http://dx.doi.org/10.1136/ (MIA) is a severe, fatal neonatal disorder, involving the no primary aetiology has been proved for the con- jmedgenet-2012-101483). occurrence of obstructions in the small and large dition. Importantly, in some cases MIA is also asso- 1 intestines ultimately leading to organ failure. Surgical ciated with either mild or severe combined Centre de Recherche du CHU fi 2–5 Ste-Justine, University of interventions are palliative but do not provide long-term immunode ciency (SCID), raising the possibility Montreal, Montreal, Quebec, survival. Severe immunodeficiency may be associated that an abnormal immune response might be the Canada with the phenotype. -

How Shelterin Protects Mammalian Telomeres 303 ANRV361-GE42-15 ARI 3 October 2008 10:10 (See 3' 5' TRF2 S Mplex
ANRV361-GE42-15 ARI 3 October 2008 10:10 ANNUAL How Shelterin Protects REVIEWS Further Click here for quick links to Annual Reviews content online, Mammalian Telomeres including: • Other articles in this volume 1 • Top cited articles Wilhelm Palm and Titia de Lange • Top downloaded articles • Our comprehensive search Laboratory for Cell Biology and Genetics, The Rockefeller University, New York, NY 10021; email: [email protected] 1Current address: Max Planck Institute of Molecular Cell Biology and Genetics, 01307 Dresden, Germany Annu. Rev. Genet. 2008. 42:301–34 Key Words First published online as a Review in Advance on ATM, ATR, cancer, NHEJ, HR August 4, 2008 by Rockefeller University on 10/13/09. For personal use only. The Annual Review of Genetics is online at Abstract genet.annualreviews.org The genomes of prokaryotes and eukaryotic organelles are usually cir- This article’s doi: cular as are most plasmids and viral genomes. In contrast, the nuclear Annu. Rev. Genet. 2008.42:301-334. Downloaded from arjournals.annualreviews.org 10.1146/annurev.genet.41.110306.130350 genomes of eukaryotes are organized on linear chromosomes, which Copyright c 2008 by Annual Reviews. require mechanisms to protect and replicate DNA ends. Eukaryotes All rights reserved navigate these problemswith the advent of telomeres, protective nucle- 0066-4197/08/1201-0301$20.00 oprotein complexes at the ends of linear chromosomes, and telomerase, the enzyme that maintains the DNA in these structures. Mammalian telomeres contain a specific protein complex, shelterin, that functions to protect chromosome ends from all aspects of the DNA damage re- sponse and regulates telomere maintenance by telomerase. -
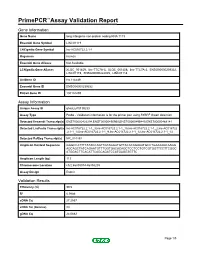
Primepcr™Assay Validation Report
PrimePCR™Assay Validation Report Gene Information Gene Name long intergenic non-protein coding RNA 1119 Ensembl Gene Symbol LINC01119 LNCipedia Gene Symbol lnc-AC016722.2.1-1 Organism Human Ensembl Gene Aliases Not Available LCNipedia Gene Aliases XLOC_001459, linc-TTC7A-3, XLOC_001458, linc-TTC7A-2, ENSG00000239332, LINC01119, ENSG00000222005, LINC01118 UniGene ID Hs.114449 Ensembl Gene ID ENSG00000239332 Entrez Gene ID 100134259 Assay Information Unique Assay ID qhsaLEP0139233 Assay Type Probe - Validation information is for the primer pair using SYBR® Green detection Detected Ensembl Transcript(s) ENST00000422294,ENST00000490950,ENST00000495449,ENST00000468141 Detected LncPedia Transcript(s) lnc-AC016722.2.1-1_3,lnc-AC016722.2.1-1_15,lnc-AC016722.2.1-1_2,lnc-AC016722 .2.1-1_14,lnc-AC016722.2.1-1_9,lnc-AC016722.2.1-1_12,lnc-AC016722.2.1-1_13 Detected RefSeq Transcript(s) NR_024452 Amplicon Context Sequence GGGCCATTTTATACCAGTTGTAGGATGTTACACAGAAATGCCTGAAAAGCAAGG AGCAGCTATCAGAATGTTTGGTGACACAGCTCCTCCTGTCGTGGTTCCTTCGGC ATGGACTTCACATTCAGCAGATCCATGAGGTGTTC Amplicon Length (bp) 113 Chromosome Location chr2:46855974-46858205 Assay Design Exonic Validation Results Efficiency (%) 99.5 R2 0.9986 cDNA Cq 27.2537 cDNA Tm (Celsius) 83 gDNA Cq 24.0682 Page 1/5 PrimePCR™Assay Validation Report Specificity (%) 100 Information to assist with data interpretation is provided at the end of this report. Page 2/5 PrimePCR™Assay Validation Report LINC01119, Human Amplification Plot Amplification of cDNA generated from 25 ng of universal reference RNA Melt Peak -

Pan-Cancer Analysis of Alternative Lengthening of Telomere Activity
cancers Article Pan-Cancer Analysis of Alternative Lengthening of Telomere Activity Ji-Yong Sung 1,2, Hee-Woong Lim 3, Je-Gun Joung 1,* and Woong-Yang Park 1,2,4,* 1 Samsung Genome Institute, Samsung Medical Center, Seoul 06351, Korea; [email protected] 2 Department of Health Science and Technology, Samsung Advanced Institute of Health Science and Technology, Sungkyunkwan University, Seoul 06351, Korea 3 Division of Biomedical Informatics, Cincinnati Children’s Hospital Medical Center, Cincinnati, OH 45229, USA; [email protected] 4 Department of Molecular Cell Biology, School of Medicine, Sungkyunkwan University, Seoul 06351, Korea * Correspondence: [email protected] (J.-G.J.); [email protected] (W.-Y.P.); Tel.: +82-2-3410-1706 (J.-G.J.); +82-2-3410-6128 (W.-Y.P.); Fax: +82-2-2148-9819 (W.-Y.P.) Received: 30 June 2020; Accepted: 5 August 2020; Published: 7 August 2020 Abstract: Alternative lengthening of telomeres (ALT) is a telomerase-independent mechanism that extends telomeres in cancer cells. It influences tumorigenesis and patient survival. Despite the clinical significance of ALT in tumors, the manner in which ALT is activated and influences prognostic outcomes in distinct cancer types is unclear. In this work, we profiled distinct telomere maintenance mechanisms (TMMs) using 8953 transcriptomes of 31 different cancer types from The Cancer Genome Atlas (TCGA). Our results demonstrated that approximately 29% of cancer types display high ALT activity with low telomerase activity in the telomere-lengthening group. Among the distinct ALT mechanisms, homologous recombination was frequently observed in sarcoma, adrenocortical carcinoma, and kidney chromophobe. Five cancer types showed a significant difference in survival in the presence of high ALT activity. -

Ailanthone Inhibits Non-Small Cell Lung Cancer Cell Growth Through Repressing DNA Replication Via Downregulating RPA1
FULL PAPER British Journal of Cancer (2017) 117, 1621–1630 | doi: 10.1038/bjc.2017.319 Keywords: ailanthone; non-small cell lung cancer; DNA replication; RPA1; Chinese medicine Ailanthone inhibits non-small cell lung cancer cell growth through repressing DNA replication via downregulating RPA1 Zhongya Ni1, Chao Yao1, Xiaowen Zhu1, Chenyuan Gong1, Zihang Xu2, Lixin Wang3, Suyun Li4, Chunpu Zou2 and Shiguo Zhu*,1,3 1Laboratory of Integrative Medicine, School of Basic Medical Sciences, Shanghai University of Traditional Chinese Medicine, 1200 Cai Lun Rd, Shanghai 201203, PR China; 2Department of Internal Classic of Medicine, School of Basic Medical Sciences, Shanghai University of Traditional Chinese Medicine, 1200 Cai Lun Rd, Shanghai 201203, PR China; 3Department of Immunology and Pathogenic Biology, School of Basic Medical Sciences, Shanghai University of Traditional Chinese Medicine, 1200 Cai Lun Rd, Shanghai 201203, PR China and 4Department of Pathology, School of Basic Medical Sciences, Shanghai University of Traditional Chinese Medicine, 1200 Cai Lun Rd, Shanghai 201203, PR China Background: The identification of bioactive compounds from Chinese medicine plays a crucial role in the development of novel reagents against non-small cell lung cancer (NSCLC). Methods: High throughput screening assay and analyses of cell growth, cell cycle, apoptosis, cDNA microarray, BrdU incorporation and gene expression were performed. Results: Ailanthone (Aila) suppressed NSCLC cell growth and colony formation in vitro and inhibited NSCLC tumour growth in subcutaneously xenografted and orthotopic lung tumour models, leading to prolonged survival of tumour-bearing mice. Moreover, Aila induced cell cycle arrest in a dose-independent manner but did not induce apoptosis in all NSCLC cells. -

A Computational Approach for Defining a Signature of Β-Cell Golgi Stress in Diabetes Mellitus
Page 1 of 781 Diabetes A Computational Approach for Defining a Signature of β-Cell Golgi Stress in Diabetes Mellitus Robert N. Bone1,6,7, Olufunmilola Oyebamiji2, Sayali Talware2, Sharmila Selvaraj2, Preethi Krishnan3,6, Farooq Syed1,6,7, Huanmei Wu2, Carmella Evans-Molina 1,3,4,5,6,7,8* Departments of 1Pediatrics, 3Medicine, 4Anatomy, Cell Biology & Physiology, 5Biochemistry & Molecular Biology, the 6Center for Diabetes & Metabolic Diseases, and the 7Herman B. Wells Center for Pediatric Research, Indiana University School of Medicine, Indianapolis, IN 46202; 2Department of BioHealth Informatics, Indiana University-Purdue University Indianapolis, Indianapolis, IN, 46202; 8Roudebush VA Medical Center, Indianapolis, IN 46202. *Corresponding Author(s): Carmella Evans-Molina, MD, PhD ([email protected]) Indiana University School of Medicine, 635 Barnhill Drive, MS 2031A, Indianapolis, IN 46202, Telephone: (317) 274-4145, Fax (317) 274-4107 Running Title: Golgi Stress Response in Diabetes Word Count: 4358 Number of Figures: 6 Keywords: Golgi apparatus stress, Islets, β cell, Type 1 diabetes, Type 2 diabetes 1 Diabetes Publish Ahead of Print, published online August 20, 2020 Diabetes Page 2 of 781 ABSTRACT The Golgi apparatus (GA) is an important site of insulin processing and granule maturation, but whether GA organelle dysfunction and GA stress are present in the diabetic β-cell has not been tested. We utilized an informatics-based approach to develop a transcriptional signature of β-cell GA stress using existing RNA sequencing and microarray datasets generated using human islets from donors with diabetes and islets where type 1(T1D) and type 2 diabetes (T2D) had been modeled ex vivo. To narrow our results to GA-specific genes, we applied a filter set of 1,030 genes accepted as GA associated. -

1 Metabolic Dysfunction Is Restricted to the Sciatic Nerve in Experimental
Page 1 of 255 Diabetes Metabolic dysfunction is restricted to the sciatic nerve in experimental diabetic neuropathy Oliver J. Freeman1,2, Richard D. Unwin2,3, Andrew W. Dowsey2,3, Paul Begley2,3, Sumia Ali1, Katherine A. Hollywood2,3, Nitin Rustogi2,3, Rasmus S. Petersen1, Warwick B. Dunn2,3†, Garth J.S. Cooper2,3,4,5* & Natalie J. Gardiner1* 1 Faculty of Life Sciences, University of Manchester, UK 2 Centre for Advanced Discovery and Experimental Therapeutics (CADET), Central Manchester University Hospitals NHS Foundation Trust, Manchester Academic Health Sciences Centre, Manchester, UK 3 Centre for Endocrinology and Diabetes, Institute of Human Development, Faculty of Medical and Human Sciences, University of Manchester, UK 4 School of Biological Sciences, University of Auckland, New Zealand 5 Department of Pharmacology, Medical Sciences Division, University of Oxford, UK † Present address: School of Biosciences, University of Birmingham, UK *Joint corresponding authors: Natalie J. Gardiner and Garth J.S. Cooper Email: [email protected]; [email protected] Address: University of Manchester, AV Hill Building, Oxford Road, Manchester, M13 9PT, United Kingdom Telephone: +44 161 275 5768; +44 161 701 0240 Word count: 4,490 Number of tables: 1, Number of figures: 6 Running title: Metabolic dysfunction in diabetic neuropathy 1 Diabetes Publish Ahead of Print, published online October 15, 2015 Diabetes Page 2 of 255 Abstract High glucose levels in the peripheral nervous system (PNS) have been implicated in the pathogenesis of diabetic neuropathy (DN). However our understanding of the molecular mechanisms which cause the marked distal pathology is incomplete. Here we performed a comprehensive, system-wide analysis of the PNS of a rodent model of DN. -

4-6 Weeks Old Female C57BL/6 Mice Obtained from Jackson Labs Were Used for Cell Isolation
Methods Mice: 4-6 weeks old female C57BL/6 mice obtained from Jackson labs were used for cell isolation. Female Foxp3-IRES-GFP reporter mice (1), backcrossed to B6/C57 background for 10 generations, were used for the isolation of naïve CD4 and naïve CD8 cells for the RNAseq experiments. The mice were housed in pathogen-free animal facility in the La Jolla Institute for Allergy and Immunology and were used according to protocols approved by the Institutional Animal Care and use Committee. Preparation of cells: Subsets of thymocytes were isolated by cell sorting as previously described (2), after cell surface staining using CD4 (GK1.5), CD8 (53-6.7), CD3ε (145- 2C11), CD24 (M1/69) (all from Biolegend). DP cells: CD4+CD8 int/hi; CD4 SP cells: CD4CD3 hi, CD24 int/lo; CD8 SP cells: CD8 int/hi CD4 CD3 hi, CD24 int/lo (Fig S2). Peripheral subsets were isolated after pooling spleen and lymph nodes. T cells were enriched by negative isolation using Dynabeads (Dynabeads untouched mouse T cells, 11413D, Invitrogen). After surface staining for CD4 (GK1.5), CD8 (53-6.7), CD62L (MEL-14), CD25 (PC61) and CD44 (IM7), naïve CD4+CD62L hiCD25-CD44lo and naïve CD8+CD62L hiCD25-CD44lo were obtained by sorting (BD FACS Aria). Additionally, for the RNAseq experiments, CD4 and CD8 naïve cells were isolated by sorting T cells from the Foxp3- IRES-GFP mice: CD4+CD62LhiCD25–CD44lo GFP(FOXP3)– and CD8+CD62LhiCD25– CD44lo GFP(FOXP3)– (antibodies were from Biolegend). In some cases, naïve CD4 cells were cultured in vitro under Th1 or Th2 polarizing conditions (3, 4). -

Liver Glucose Metabolism in Humans
Biosci. Rep. (2016) / 36 / art:e00416 / doi 10.1042/BSR20160385 Liver glucose metabolism in humans Mar´ıa M. Adeva-Andany*1, Noemi Perez-Felpete*,´ Carlos Fernandez-Fern´ andez*,´ Cristobal´ Donapetry-Garc´ıa* and Cristina Pazos-Garc´ıa* *Nephrology Division, Hospital General Juan Cardona, c/ Pardo Bazan´ s/n, 15406 Ferrol, Spain Synopsis Information about normal hepatic glucose metabolism may help to understand pathogenic mechanisms underlying obesity and diabetes mellitus. In addition, liver glucose metabolism is involved in glycosylation reactions and con- nected with fatty acid metabolism. The liver receives dietary carbohydrates directly from the intestine via the portal vein. Glucokinase phosphorylates glucose to glucose 6-phosphate inside the hepatocyte, ensuring that an adequate flow of glucose enters the cell to be metabolized. Glucose 6-phosphate may proceed to several metabolic path- ways. During the post-prandial period, most glucose 6-phosphate is used to synthesize glycogen via the formation of glucose 1-phosphate and UDP–glucose. Minor amounts of UDP–glucose are used to form UDP–glucuronate and UDP– galactose, which are donors of monosaccharide units used in glycosylation. A second pathway of glucose 6-phosphate metabolism is the formation of fructose 6-phosphate, which may either start the hexosamine pathway to produce UDP-N-acetylglucosamine or follow the glycolytic pathway to generate pyruvate and then acetyl-CoA. Acetyl-CoA may enter the tricarboxylic acid (TCA) cycle to be oxidized or may be exported to the cytosol to synthesize fatty acids, when excess glucose is present within the hepatocyte. Finally, glucose 6-phosphate may produce NADPH and ribose 5-phosphate through the pentose phosphate pathway. -

Dyskerin Mutations Present in Dyskeratosis Congenita Patients Increase Oxidative Stress and DNA Damage Signalling in Dictyostelium Discoideum
cells Article Dyskerin Mutations Present in Dyskeratosis Congenita Patients Increase Oxidative Stress and DNA Damage Signalling in Dictyostelium Discoideum Javier Rodriguez-Centeno, Rosario Perona and Leandro Sastre * Instituto de Investigaciones Biomedicas, CSIC/UAM and Centro de Investigación Biomédica en Red de Enfermedades Raras, CIBERER, 28029 Madrid, Spain; [email protected] (J.R.-C.); [email protected] (R.P.) * Correspondence: [email protected]; Tel.: +3491-585-4437 Received: 11 October 2019; Accepted: 5 November 2019; Published: 8 November 2019 Abstract: Dyskerin is a protein involved in the formation of small nucleolar and small Cajal body ribonucleoproteins. These complexes participate in RNA pseudouridylation and are also components of the telomerase complex required for telomere elongation. Dyskerin mutations cause a rare disease, X-linked dyskeratosis congenita, with no curative treatment. The social amoeba Dictyostelium discoideum contains a gene coding for a dyskerin homologous protein. In this article D. discoideum mutant strains that have mutations corresponding to mutations found in dyskeratosis congenita patients are described. The phenotype of the mutant strains has been studied and no alterations were observed in pseudouridylation activity and telomere structure. Mutant strains showed increased proliferation on liquid culture but reduced growth feeding on bacteria. The results obtained indicated the existence of increased DNA damage response and reactive oxygen species, as also reported in human Dyskeratosis congenita cells and some other disease models. These data, together with the haploid character of D. discoideum vegetative cells, that resemble the genomic structure of the human dyskerin gene, located in the X chromosome, support the conclusion that D. discoideum can be a good model system for the study of this disease. -

The Capacity of Long-Term in Vitro Proliferation of Acute Myeloid
The Capacity of Long-Term in Vitro Proliferation of Acute Myeloid Leukemia Cells Supported Only by Exogenous Cytokines Is Associated with a Patient Subset with Adverse Outcome Annette K. Brenner, Elise Aasebø, Maria Hernandez-Valladares, Frode Selheim, Frode Berven, Ida-Sofie Grønningsæter, Sushma Bartaula-Brevik and Øystein Bruserud Supplementary Material S2 of S31 Table S1. Detailed information about the 68 AML patients included in the study. # of blasts Viability Proliferation Cytokine Viable cells Change in ID Gender Age Etiology FAB Cytogenetics Mutations CD34 Colonies (109/L) (%) 48 h (cpm) secretion (106) 5 weeks phenotype 1 M 42 de novo 241 M2 normal Flt3 pos 31.0 3848 low 0.24 7 yes 2 M 82 MF 12.4 M2 t(9;22) wt pos 81.6 74,686 low 1.43 969 yes 3 F 49 CML/relapse 149 M2 complex n.d. pos 26.2 3472 low 0.08 n.d. no 4 M 33 de novo 62.0 M2 normal wt pos 67.5 6206 low 0.08 6.5 no 5 M 71 relapse 91.0 M4 normal NPM1 pos 63.5 21,331 low 0.17 n.d. yes 6 M 83 de novo 109 M1 n.d. wt pos 19.1 8764 low 1.65 693 no 7 F 77 MDS 26.4 M1 normal wt pos 89.4 53,799 high 3.43 2746 no 8 M 46 de novo 26.9 M1 normal NPM1 n.d. n.d. 3472 low 1.56 n.d. no 9 M 68 MF 50.8 M4 normal D835 pos 69.4 1640 low 0.08 n.d. -

POLE2 Knockdown Reduce Tumorigenesis in Esophageal Squamous Cells Yongjun Zhu, Gang Chen, Yang Song, Zhiming Chen* and Xiaofeng Chen*
Zhu et al. Cancer Cell Int (2020) 20:388 https://doi.org/10.1186/s12935-020-01477-4 Cancer Cell International PRIMARY RESEARCH Open Access POLE2 knockdown reduce tumorigenesis in esophageal squamous cells Yongjun Zhu, Gang Chen, Yang Song, Zhiming Chen* and Xiaofeng Chen* Abstract Background: Esophageal squamous cell carcinoma (ESCC) is one of the most frequent malignant tumors originated from digestive system around the world and the treatment was limited by the unclear mechanism. DNA polymerase epsilon 2, accessory subunit (POLE2) is involved in DNA replication, repair, and cell cycle control, whose association with ESCC is still not clear. Methods: In this study, the expression level of POLE2 in ESCC tissues was detected by IHC. The POLE2 knockdown cell line was constructed, identifed by qPCR and western blot and used for detecting cellular functions and con- structing xenotransplantation mice model. MTT Assay, colony formation assay, fow cytometry, wound-healing assay and Transwell assay were used to detected cell proliferation, apoptosis and migration. Results: We frstly identifed that the expression of POLE2 was overexpressed in ESCC. Moreover, the high expres- sion of POLE2 can predict the tumor deterioration and poor prognosis of ESCC patients. Additionally, downregulation of POLE2 was involved in ESCC progression by promoting proliferation, migration, and inhibiting apoptosis in vitro. In vivo studies proved that POLE2 was positively correlated with ESCC tumor formation, which was consistent with the results in vitro. We also illuminated that POLE2 knockdown upregulated pro-apoptotic proteins (Bax, Caspase3, CD40L, FasL, IGFBP-5 and P21) and downregulated anti-apoptotic proteins (CLAP-2, IGF-I and sTNF-R2).