Airborne and Deposited Radioactivity in Finland in 1987
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-
Savonlinna ENG.Pdf
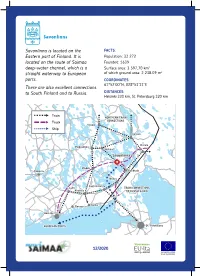
Savonlinna is located on the FACTS: Eastern part of Finland. It is Population: 33 272 located on the route of Saimaa Founded: 1639 deep-water channel, which is a Surface area: 3 597,70 km2 straight waterway to European of which ground area: 2 238.09 m2 ports. COORDINATES: 61°52’00”N, 028°53’11”E There are also excellent connections to South Finland and to Russia. DISTANCES: Helsinki 330 km, St. Petersburg 320 km Train NORTHERN TRAIN CONNECTIONS Truck Vaasa Kuopio Ship Joensuu Niirala Pieksämäki Varkaus Savonlinna Mikkeli Tampere Parikkala Pori Imatra Lappeenranta Lahti TRAIN CONNECTIONS Kouvola TO RUSSIA & ASIA Kotka Tur ku Porvoo Helsinki EUROPEAN PORTS St. Petersburg 12/2020 PORT OF VUOHISAARI Number of Berths: PORT SERVICES: 1, lenght of berth 100 m Savonlinnan kaupunki Maximum size of vessels: Port Manager Jyrki Haajanen • Lenght: 82,5 m +358 (0) 44 417 4622 • Width: 12,6 m [email protected] • Draft: 4,35 m Vuohisaarenkatu 5 Services: 57230 SAVONLINNA • Electricity • Water STEVEDORING & FORWARDING: • Bilge • Septic tank station Oy Saimaa Terminals Ab Yhteydet: Hannu Kaipainen • Good access by rail and road +358 (0) 50 327 0341 (Main road 14) [email protected] www.sterm.fi GUEST MARINAS PASSENGER PORT There are 3 guest marinas in the city The Passenger Port of Savonlinna is one center of Savonlinna and 1 in Savonranta. of the vivacious passenger ports of Lake All guest marinas have shower, toilet and Saimaa area. It is located in the city center restaurant services. and is surrounded by excellent Hotel and Restaurant services. The local market Guest marina of Spahotel Casino square is located next to the port. -

Regional Sessions of EYP Finland Information Sheet for Officials
t Regional Sessions of EYP Finland Information Sheet for Officials Helsinki | 24–26 October Savoninna | 31 October–2 November Salo | 7–9 November Oulu | 14–16 November 1 Welcome to the Regional Sessions! Helsinki 2014 European Youth Parliament Finland will organise its annual Regional Sessions for 24–26 October the seventh time in 2014. The sessions will take place in Helsinki, Savonlinna, Salo and Oulu in October and November. The Regional Sessions act as a preliminary Head-organised by Katariina round for the 21st National Session of EYP Finland, to be held in Turku in January Knuuttila, Patrik Kumpulainen 2015. and Viktor Salenius. Since 2008, the Regional Sessions of EYP Finland have been a gateway to the world Savonlinna of EYP for thousands of high school students in Finland. They have also gained a 31 October–2 November solid reputation as high-quality events among the international EYP community as indicated by the high number of applications for officials’ posts in the recent years. Head-organised by Essi Hy- With organising four sessions for the second year now, we are offering a total of vönen and Sara Välimäki. almost a hundred officials’ posts! Salo EYP Finland now welcomes you all to apply for another round of Regional Sessions 7–9 November in Finland. The call is open for vice-presidents, editors, chairs and journalists. The sessions in Helsinki and Salo will each host a team of 17 chairs, eight journalists and Head-organised by Niko Lammi, 1–2 editors; the sessions in Savonlinna and Oulu will have a team of 12 chairs, six Laura Uusitalo and Riku Suvi- journalists and 1–2 editors. -

The Saima District
THE SAIMA DISTRICT SAVONLINNA-KUOPIO "THE PKftRL OF SrtIJVM" THE LEPPÄVIRTA ROUTE EDITED BY THE KUOPIO SECTION Ul' THE FINNISH TOURIST ASSOCIATION. THE SAIMA DISTRICT SAVONLINNA-KUOPIO THE LEPPÄVIRTA ROUTE EDITED BY THE KUOPIO SECTION OF THE FINNISH TOURIST ASSOCIATION PRINTED IN KUOPIO 1928 BY POHJOIS-SAVON KIRJAPAINO O.Y 2 The harbour of Savonlinna TJ*inland does not offer the foreign tourist much in the way of historic monuments or famous art treasures, but she offers so much more in the way of beautiful scenery, the peace and pleasure of which comes from being in close contact with untouched nature. The beauty of the country lies especially in the extensive forests, and in the sin- gular profusion of lakes. It is most apparent in the large lake district, and in the magnificent wa- 3 terways of Lake Saima. Here the admirer of na~ fure and the tourist receives a never to be forgot- ten impression. Here the tourist, who has visited different parts of the country may exclaim: „The chain of the Saima lakes is the most wonderful thing I have ever seen; here is the northern para- dise of hills, forests, and lakes." But as a boat trip along this chain of lakes, with its many branches extending for miles in all directions, demands too much of the tourist's pre- cious time, he must satisfy himself by experiencing the best part of all by undertaking the journey from Savonlinna via Varkaus to Kuopio, from the border of Olof's Castle to the base of the stately Puijo Hill. -

Ita-Suomi FI13 RIM Regional Innovation Report
Version: Final Date: 19 August 2011 Regional Innovation Monitor Regional Innovation Report (Eastern Finland/Itä-Suomi) To the European Commission Enterprise and Industry Directorate-General Directorate D – Industrial Innovation and Mobility Industries Kimmo Viljamaa Henri Lahtinen Advansis www.technopolis-group.com PREFACE The Regional Innovation Monitor (RIM)1 is an initiative of the European Commission's Directorate General for Enterprise and Industry, which has the objective to describe and analyse innovation policy trends across EU regions. RIM analysis is based on methodologies developed in the context of the INNO-Policy Trendchart which covers innovation policies at national level as part of the PRO INNO Europe initiative. The overarching objective of this project is to enhance the competitiveness of European regions through increasing the effectiveness of their innovation policies and strategies. The specific objective of the RIM is to enhance the scope and quality of policy assessment by providing policy-makers, other innovation stakeholders with the analytical framework and tools for evaluating the strengths and weaknesses of regional policies and regional innovation systems. RIM covers EU-20 Member States: Austria, Belgium, Bulgaria, the Czech Republic, Denmark, Finland, France, Germany, Greece, Hungary, Ireland, Italy, the Netherlands, Poland, Portugal, Romania, Slovakia, Spain, Sweden and the United Kingdom. This means that RIM will not concentrate on Member States where the Nomenclature of territorial units for statistics NUTS 1 and 2 levels are identical with the entire country (Estonia, Latvia, and Lithuania), Malta which only has NUTS 3 regions, Slovenia which has a national innovation policy or Cyprus and Luxembourg which are countries without NUTS regions. -

Soile Yli-Mäyry Resume 2012 WWG
W A L T E R W I C K I S E R G A L L E R Y I N C . 210 Eleventh Avenue Suite 303 New York, NY 10001 T 212-941-1817 F 212-625-0601 [email protected] www.walterwickisergallery.com Soile Yli-MÄyry Selected Solo Exhibitions 2011 Kelley Roy Gallery, Miami, Florida Gallery Viktoria, Gothenburg, Sweden Walter Wickiser Gallery, New York Gallery Seira, Tokyo, Japan Soile Yli-Mayry Art Hall, Kuortane, Finland Gallery D’ Arte II Saggiatore, Rome, Italy Artfolio Gallery, Singapore Gallery Etienne de Causans, Paris, France 2010 Art Chamber, Goa, India Art Chicago, Chicago Matsuya Ginza Gallery, Tokyo, Japan Soile Yli-Mayry Art Hall, Kuortane, Finland Toronto International Art Fair, Toronto, Canada Gallery Arktika, Marktoberdorf, Germany State of-the-Arts Gallery, Hong Kong Art Miami, Miami 2009 Kelley Roy Gallery, Miami, Florida Walter Wickiser Gallery, New York Ofoten Museum, Narvik, Norway Lakshana Art Gallery, Chennai, India Gallery Tobias Loeffel, Basel, Switzerland Gallery Tobias Loeffel, Luzern, Switzerland Soile Yli-Mayry Art Hall, Kuortane, Finland Moisio Art Manor, Kouvola, Finland Scand Art Gallery, Hollviken, Sweden 2008 Gallery Etienne de Causans, Paris, France China Art Museum, Beijing, China Green Art Gallery, Dubai, United Arab Emirates Walter Wicksier Gallery | 210 Eleventh Avenue Suite 303 | T: 212-941-1817 | [email protected] Matsuya Ginza Gallery, Tokyo, Japan Soile Yli-Mäyry Art Hall, Kuortane, Finland 2007 Art Chamber, Goa, India Walter Wickiser Gallery, New York Gallery Nievergelt, Zurich, Switzerland Gallery Port, Ginza, Tokyo, -

Merimieslääkärintarkast
MERIMIESLÄÄKÄRINTARKASTUS Sinut on hyväksytty opiskelemaan merenkulkua Kaakkois-Suomen ammattikorkeakouluun. Merenkulkualalla ammatilliset pätevyys- ja terveysvaatimukset on määritelty tavanomaista tarkemmin kansainvälisin sopimuksin ja kansallisella lainsäädännöllä. Näin ollen merenkulkualan koulutusta antaviin oppilaitoksiin hyväksytyltä vaaditaan terveydentilasta hyväksytty merimieslääkärintodistus. Normaalitilanteissa lääkärintodistus on voimassa kaksi vuotta. Sinulle on suoritettava alkutarkastus merimieslääkärin tekemänä, merimiesterveyskeskuksessa. Voit hankkia todistuksen joko omalta paikkakunnaltasi tai paikkakuntaasi lähimpänä olevasta meri- miesterveyskeskuksesta. Huomioithan, että kaikki merimieslääkäripalveluja tuottavat yksiköt eivät ole merimiesterveyskeskuksia. Merimiesterveyskeskuksia on seuraavissa kaupungeissa: Hamina, Helsinki, Kemi, Kotka, Oulu, Pietarsaari, Pori, Rauma, Savonlinna, Turku ja Vaasa. Katso tarkempi lista merimiesterveyskeskuksista tämän tiedotteen liitteestä (liite 1). Laivaväen terveysvaatimuksista voit käydä lukemassa lisää esimerkiksi Työterveyslaitoksen sivuilta (www.ttl.fi/laivavaen-laakarintarkastukset). Merimieslääkärintodistus tarkastetaan opintojesi alussa orientaatioviikolla. Sinun tulee esittää todistus merenkulun assistentti Outi Hemmilälle. Muina aikoina voit sopia erillisen ajan sähköpostitse [email protected]. Merimieslääkärintodistus tulee esittää 15.9.2021 mennessä, muutoin opiskeluoikeus voidaan purkaa. Liite 1 Merimiesterveyskeskukset, yhteystiedot 13.4.2021 HAMINA: Haminassa laivaväen -

Savonlinna Euroopan Kulttuuripääkaupungiksi 2026 -Hanke
Saimaa-ilmiöllä elinvoimaa Järvi-Suomeen - Savonlinna Euroopan kulttuuripääkaupungiksi 2026 -hanke Tausta Euroopan kulttuuripääkaupunkeja (European Capital of Culture, ECoC) on nimetty vuodesta 1985. Suomesta kulttuuripääkaupunkeina ovat olleet aiemmin Helsinki vuonna 2000 ja Turku vuonna 2011. Vuonna 2026 Euroopan kulttuuripääkaupungit tulevat Suomesta ja Slovakiasta. Etelä-Savon maakuntaliitto hallinnoi yhteistyöhanketta ”Kulttuurista elinvoimaa Järvi-Suomeen – Savonlinna Euroopan kulttuuripääkaupungiksi 2026” ajalla 1.3.2019 – 31.8.2020. Hankkeen tuella Savonlinnan, Joensuun, Kuopion, Lappeenrannan ja Mikkelin kaupungit sekä Pohjois-Karjalan, Pohjois- Savon, Etelä-Karjalan ja Etelä-Savon maakuntaliitot tavoittelevat Savonlinnan valintaa Euroopan kulttuuripääkaupungiksi vuodeksi 2026. Savonlinna on hankkeen yhteisesti nimetty ydinkaupunki, koska hakemus jätetään menettelyohjeen (EU/OKM) mukaan yhden kaupungin nimissä. Ohjelma levittäytyy kuitenkin kaikkiin em. kaupunkeihin ja maakuntiin. Ohjelmassa tehtävät toimenpiteet jakautuvat suhteessa kaupunkien omarahoitusosuuksiin kaupunkikohtaiseksi. Kulttuuripääkaupunkihausta käytetään nimitystä Saimaa-ilmiö2026. Euroopan kulttuuripääkaupunkihaku on kaksivaiheinen prosessi. Ensimmäisen vaiheen hakemus, joka käytännössä on 60-sivuinen kirja, tulee jättää Opetus- ja kulttuuriministeriölle 5. toukokuuta 2020 mennessä. Tämä hakemus, hakukirja valmistellaan em. hankkeen puitteissa. Toiseen valintavaiheeseen pääsevät kaupungit julkistetaan valintaraadin esivalintakokouksen päätteeksi Helsingissä -

Bigger Picture
BUILDING THE BIGGER PICTURE ANNUAL REPORT 2011 10 January 2011 16 February 2011 23 February 2011 3 May 2011 25 May 2011 17 August 2011 30 August 2011 15 September 2011 4 October 2011 Destia and Ramirent Destia wins a prize in the Destia participates in the Natural gas Civil engineering Agreement between Destia Main Road 51 bridge Oikarainen Bridge Destia highly commended in the enter co-operation Uusimaa occupational Liikennerevoluutio (traffic pipeline laid in contract for Destia at and the City of Vantaa for moving work begins completed and Environmental Structure of the Year safety competition revolution) ideas competition Hiidenvesi Ovako’s steel plant Tikkurilantie ST contract at Espoonlahti inaugurated competition 1 February 2011 The 21 February 2011 4 March 2011 20 April 2011 1 June 2011 4 July 2011 17 August 2011 13 September 2011 22 September 2011 7 October 2011 METRO consortium wins METRO signs a contract Destia at the Destia wins seven Area-wide contract Destia sells its Vaasa Destia’s construction Nationally significant Victoria Square in Destia gives award for the Safest Metro Site agreement for the Infra 11 event area-wide mainte- for Destia at Maaria- depot area to Kesko work at the centre of research work for traffic Tornio completed best dissertation in the competition Karhusaari metro tunnels nance contracts Paattinen in Turku Kalasatama begins and environmental planning field of infrastructure 2 2 BUILDING THE BIGGER PICTURE CONTENTS Destia in 2011 3 Destia’s business 12 Corporate responsibility 19 CEO’s review 4 Destia’s services 13 Financial responsibility 20 Destia’s strategy Infra construction 14 Social responsibility 20 2011 – 2014 6 Infra maintenance 15 Environmental Destia’s risk management 8 Railways 16 responsibility 22 Operating environment Rock 17 Organisation and and market review 10 Consulting services 18 main offices 23 30 November 2011 Destia sells its surfacing business to NCC DESTIA BUILDS, MAINTAINS AND DESIGNS TRAFFIC AND INDUSTRIAL ENVIRONMENTS, SO THAT THE LIVING ENVIRONMENT WILL BE EFFICIENT AND SAFE. -

ÄITI SAIMAA ELÄMYSMATKA 7 Vrk Matkaisäntänä Folke West
ÄITI SAIMAA ELÄMYSMATKA 7 vrk Matkaisäntänä Folke West Lappeenranta – Imatra – Parikkala – Punkaharju – Savonlinna – Järvisydän – Mikkeli Lähtö ja paluu Helsinki tai Tampere Äiti Saimaa kutsuu lähde mukaan viikon elämysmatkalla - tarjolla luonnon kauniita maisemia – huippuhotellit- mielenkiintoista historiaa, kulttuurielämyksiä ja rentouduttavaa liikuntaa! Päivä 1 Matka Lappeenrantaan Äiti Saimaa kiertueen ensimmäinen pysähdyspaikka on itseoikeutetusti maaherra Johan Rosenhanen vuonna 1639 Pien Saimaan rannalle perustama Villmanstrand eli Lappeenranta. Majoitumme Rakuuna hotelliin ja illastamme Lappeenrannan linnoituksen puolella Saimaan äärellä ravintola Wanhassa Makasiinissa. Päivä 2 Villmanstrand – Lappeenranta Kauounkiretki vie Lappeenrannan keskustaan, linnoitukseen ja satamaan. Kahvit juodaan Kehruuhuoneella ja samalla tutustutaan kotimuseoon. Illallinen nautitaan Kippusarven Säräpirtti ravintolassa. Päivä 3 Imatra – Parikkala – Punkaharju -Savonlinna Lappeenrannasta siirrymme toisiin Saimaan maisemiin Imatralle, jonka maineikas koski on kiinnostanut matkailijoita läpi vuosisatojen. 1600 luvulla rakennettiin, kosken kupeeseen ensimmäinen krouvi ja majatalo. Keisarinna Katariina II käväisi Ihailemassa koskea 126 hevosen saattueella ja vierailun seurauksena alue julistettiin 1741 luonnonpuistoksi. Julkkisvierailijoita riitti myöhemminkin, heidän joukossaan kirjailija Alexandre Dumas ja säveltäjä Richard Wagner. Nykyinen majoituspaikka, komea jugend tyylinen Valtionhotelli valmistui vuonna 1903 ja toimii ansiokkaasti tänäkin päivänä. -

Savonlinnan Kaupungin Sote-Alueen Valmistelusuunta
Savonlinnan kaupungin sote-alueen valmistelusuunta Vastaajien kokonaismäärä: 341 1. Kommentti tai kannanotto Vastaajien määrä: 341 Vastaukset Ainut ja oikea suunta on Kuopio, Mikkelistä saamme pelkkää kurjuutta Ehdottomasti yhteistyöhön Pohjois-Savon ja KYS:n kanssa. Keinotekoisen Etelä-Savon maakunnan tekohengittäminen sote-yhteistyöllä Essoten kanssa on ajan ja rahan haastausta. Kannatan Pohjois-Savon suuntaa. Perusteluina, että jo nyt teemme paljon yhteistyötä Kuopion yliopistollisen sairaalan kanssa. Jos suunta on Pohjois-Savo, niin ammattikorkeakoulun tulevaisuus pitää turvata ja samoin sen hanketoiminta täällä Savonlinnassa. Eli jos suuntaamme sotessa Pohjois-Savoon, niin joudummeko vaihtamaan maakuntaa jne. Mitä se sitten tarkoittaisi ammattikorkeakoulun ja hanketoiminnan näkökulmasta? Kuopion suuntaan kiitos, aina parempi olla isomman sairaalan alla. Eikä varsinaisesti haittaakaan että kyseessä yliopistollinen sairaala. Kuopion suunta on oikea suunta. Kuopioon. Kuopion suuntaan erikoissairaanhoitoon. Paras ratkaisu olisi Mikkelin ja Savonlinnan tasavertainen yhteistyö ja työnjako. Ei uskoisi olevan mahdotonta, että lääkärit kävisivät töissä molemmissa sairaaloissa. Suurissa kaupungeissa työmatkat ovat usein 1 - 1,5 tuntia, joka on ajoaika Mikkelin ja Savonlinnan välillä. Täällä matkaa mitataan kilometreissä, kun mittayksikön tulisi olla matkaan käytetty aika. Jos edellinen ratkaisu ei ole mahdollinen tai siihen jää epävarmuustekijöitä, niin paras vaihtoehto on yhteistyö KSS kanssa. Siellä on sekä osaamista, että resursseja. Ehdottomasti -

Saimaa Lake Tour 2005
Saimaa Lake Tour 2005 This short tour takes you to the heart of the Finnish lake Prices per Person district, where you see some beautiful lake scenery, which In shared twin room € 560 the country is famous for. Visit historical landmarks and Single supplement € 110 learn about the eastern orthodox traditions on a monastery Child under 12 years in extra bed € 470 visit. sharing the room with two adults Extra bed for 3rd adult € 560 Departures June 3, 10, 17, 23, 30 Tour Includes July 6, 12, 18, 24, 30 • accommodation in twin-bedded rooms with bath or August 3, 7, 11, 15, 19 shower • luggage handling in hotels Duration • 2 buffet breakfasts, 2 lunches and 2 dinners 3 days / 2 nights • transportation by private coach • sightseeing, visits, excursions and entrance fees Meeting Point and Time in Helsinki according to the program In front of the hotel’s entrance at: Radisson SAS Plaza at • multilingual tour leader 8.15 a.m., Sokos Hotel Vaakuna 8.30, Cumulus • domestic flight Joensuu-Helsinki Seurahuone 8.30, Scandic Hotel Simonkenttä 8.45, and • service charges and taxes on included items Scandic Hotel Continental at 9.00 am. Day 1: Helsinki-Porvoo-Ruotsinpyhtää-Langinkoski-Imatra Day 2: Imatra-Punkaharju-Savonlinna-Valamo-Koli Day 3: Koli-Joensuu-Helsinki Day 1: Tracing the Czar’s Footsteps After lunch in Punkaharju you head to Savonlinna. On a (L, D, coach, 270 km / 168 miles) small island in the centre of the town stands the best- preserved medieval fortress in the Nordic countries, Your journey through the Finnish Lakeland starts as you Olavinlinna Castle. -

ANDRITZ in FINLAND FORERUNNER of INNOVATIVE TECHNOLOGIES ANDRITZ in Finland – Passion for Innovation
ANDRITZ IN FINLAND FORERUNNER OF INNOVATIVE TECHNOLOGIES ANDRITZ in Finland – passion for innovation ANDRITZ Oy is a leading global supplier of systems, equipment and services for the pulp and paper industry, including wood processing, fiber processing, chemical recovery, and stock preparation. In addition, ANDRITZ Oy offers biomass boilers and gasification plants for energy production. ANDRITZ Oy has four subsidiaries: ANDRITZ Hydro Oy, FORSEE DIGITALLY – ANDRITZ SOLUTIONS located in Tampere, delivers systems, equipment, Digitalization is a driver for growth at ANDRITZ in and services for hydraulic power generation. ANDRITZ Finland, and it continuously offers new business Fabrics and Rolls Oy located in Kerava, delivers opportunities. We help our customers to make use of world leading roll technology and Rubber Blanket for opportunities and competitive advantages by means Extensible Paper. ANDRITZ Savonlinna Works Oy is of our latest technologies. As a result, significant a manufacturing facility where key components, as improvements are achieved in the reliability and well as spare and wear parts for ANDRITZ equipment performance of the customers’ equipment, systems, and systems are produced and assembled. ANDRITZ and processes. Warkaus Works Oy is an engineering company based in Varkaus, manufacturing pressure-bearing How does ANDRITZ integrate the exciting new components for recovery boilers and power plant possibilities that come with Industry 4.0? As a tech- boilers for industrial applications. nology leader with extensive and longterm experience in supplying industrial measurement, control, and PASSION FOR INNOVATION optimization solutions for various industries, ANDRITZ ANDRITZ’s continuous research and development is combining its process and equipment expertise with activities focus on supporting customers to achieve the latest advancements in the digital era.