Atomic Structure of a Fragment of Human CD4 Containing Two Immunoglobulin-Like Domains Jiahuai Wang*, Youwei Yan*T, Thomas P
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Supplementary Table 1: Adhesion Genes Data Set
Supplementary Table 1: Adhesion genes data set PROBE Entrez Gene ID Celera Gene ID Gene_Symbol Gene_Name 160832 1 hCG201364.3 A1BG alpha-1-B glycoprotein 223658 1 hCG201364.3 A1BG alpha-1-B glycoprotein 212988 102 hCG40040.3 ADAM10 ADAM metallopeptidase domain 10 133411 4185 hCG28232.2 ADAM11 ADAM metallopeptidase domain 11 110695 8038 hCG40937.4 ADAM12 ADAM metallopeptidase domain 12 (meltrin alpha) 195222 8038 hCG40937.4 ADAM12 ADAM metallopeptidase domain 12 (meltrin alpha) 165344 8751 hCG20021.3 ADAM15 ADAM metallopeptidase domain 15 (metargidin) 189065 6868 null ADAM17 ADAM metallopeptidase domain 17 (tumor necrosis factor, alpha, converting enzyme) 108119 8728 hCG15398.4 ADAM19 ADAM metallopeptidase domain 19 (meltrin beta) 117763 8748 hCG20675.3 ADAM20 ADAM metallopeptidase domain 20 126448 8747 hCG1785634.2 ADAM21 ADAM metallopeptidase domain 21 208981 8747 hCG1785634.2|hCG2042897 ADAM21 ADAM metallopeptidase domain 21 180903 53616 hCG17212.4 ADAM22 ADAM metallopeptidase domain 22 177272 8745 hCG1811623.1 ADAM23 ADAM metallopeptidase domain 23 102384 10863 hCG1818505.1 ADAM28 ADAM metallopeptidase domain 28 119968 11086 hCG1786734.2 ADAM29 ADAM metallopeptidase domain 29 205542 11085 hCG1997196.1 ADAM30 ADAM metallopeptidase domain 30 148417 80332 hCG39255.4 ADAM33 ADAM metallopeptidase domain 33 140492 8756 hCG1789002.2 ADAM7 ADAM metallopeptidase domain 7 122603 101 hCG1816947.1 ADAM8 ADAM metallopeptidase domain 8 183965 8754 hCG1996391 ADAM9 ADAM metallopeptidase domain 9 (meltrin gamma) 129974 27299 hCG15447.3 ADAMDEC1 ADAM-like, -

The Role of Fc Gamma Receptors in the Activity of Therapeutic Monoclonal Antibodies
UNIVERSITY OF SOUTHAMPTON FACULTY OF MEDICINE Cancer Sciences Unit Volume 1 of 1 The Role of Fc Gamma Receptors in the Activity of Therapeutic Monoclonal Antibodies by Robert James Oldham Thesis for the degree of Doctor of Philosophy September 2016 UNIVERSITY OF SOUTHAMPTON ABSTRACT FACULTY OF MEDICINE Biomedicine Thesis for the degree of Doctor of Philosophy THE ROLE OF FC GAMMA RECEPTORS IN THE ACTIVITY OF THERAPEUTIC MONOCLONAL ANTIBODIES Robert James Oldham Fc gamma receptors (FcγRs) are the major family of receptors responsible for interacting with immunoglobulin G (IgG). They are known to be required for the anti-tumour activity of direct targeting mAbs through expression on NK cells and macrophages. Furthermore, recent work has suggested that cross-linking via FcγRs is required for the activity of agonistic, immune modulatory mAb. This thesis sought to investigate the requirement for these receptors for different aspects of mAb activity; from T cell activation to tumour depletion, using a combination of in vitro and in vivo systems. A panel of CHO-K1 cells were generated and transfected to express the polymorphic variants of human FcγRs. These were characterised for their ability to bind IgG before being used as feeder cells in T cell proliferation assays. The assays found that cross-linking of the anti-CD28 mAb, TGN1412 by FcγRIIb (CD32b) or FcγRIIa (CD32a) but not FcγRIIIa (CD16a) transfected cells induced T cell proliferation. Furthermore, this was accompanied by the release of pro-inflammatory cytokines including TNF-α, IFN-γ and IL-2. With the importance of cross-linking via CD32b demonstrated, experiments probed the mechanism of expression using Ramos and Raji cells. -

Cloning and Characterization of Ovine Immunoglobulin G Fc Receptor II (Fcgrii)§ Veterinary Immunology and Immunopathology
Veterinary Immunology and Immunopathology 133 (2010) 243–249 Contents lists available at ScienceDirect Veterinary Immunology and Immunopathology journal homepage: www.elsevier.com/locate/vetimm Short communication Cloning and characterization of ovine immunoglobulin G Fc receptor II (FcgRII)§ Yunchao Liu a,b, Aiping Wang b, Songlin Qiao a, Gaiping Zhang a,*, Jun Xi a, Leiming You a, Xiaohui Tian a, Qiaomu Li a, Lina Zhang a, Junqing Guo a a Key Laboratory of Animal Immunology of the Ministry of Agriculture, Henan Provincial Key Laboratory of Animal Immunology, Henan Academy of Agricultural Sciences, Zhengzhou 450002, China b College of Bioengineering, Zhengzhou University, Zhengzhou 450001, China ARTICLE INFO ABSTRACT Article history: Immunoglobulin G (IgG) Fc receptors (FcgRs) bind to immune complexes through Received 31 December 2008 interactions with the Fc region of IgG to initiate or inhibit the defense mechanism of the Received in revised form 1 July 2009 leukocytes on which they are expressed. In this study, we describe the cloning, sequencing Accepted 9 July 2009 and characterization of ovine FcgRII. By screening a translated expression sequence tag (EST) database with the protein sequence of bovine IgG Fc receptor II, we identified a Keywords: putative ovine homologue. Using rapid amplification of cDNA ends (RACE), we isolated the Sheep cDNA encoding ovine FcgRII from peripheral blood leucocyte RNA. The ovine FcgRII cDNA FcgRII contains an 894 bp open-reading frame, encoding a 297 amino acid transmembrane Inhibitory receptor Expression glycoprotein composed of two immunoglobulin-like extracellular domains, a transmem- brane region and a cytoplasmic tail with an immunoreceptor tyrosine-based inhibitory motif (ITIM). -

Molecular and Cellular Studies Examining the Biological Significance of Different Isoforms of the Receptor Tyrosine Kinase' C-Kit
Molecular and cellular studies examining the biological significance of different isoforms of the receptor tyrosine kinase' c-Kit Antony Charles Cambareri B. Sc. (Hons) Institute of Medical and Veterinary Science Department of Medicine University of Adelaide South Australia A thesis submitted to the University of Adelaide in candidature for the degree of Doctor of Philosophy October 2004 Declaration This thesis contains no material that has been accepted for the award of any other degree or diploma in any other university. To the best of my knowledge and belief' this thesis contains no material previously published or written by another person' except where due reference has been made in the text. I give consent to this copy of my thesis, when deposited in the University Library, being available for loan and photocopylng. p? Antony C atefl 4 ,(? ll Acknowledgments I would like to sincerely thank my supervisors, Professor Leonie Ashman and professor Bik To. I thank them both for their encouragement, persistence and support over the period of this work' Special thanks to Steve Fitter, who was pivotal in "retraining" me in the art of science upon my return to the lab from "the dark side". He is both a great friend and a legendary molecular biologist (it is true that it will work if you add a microlitre!)' I also thank Andrew Zannettino for spending the time to critique drafts of this thesis, and for valuable input to experimental design. I acknowledge the valuable technical assistance of Ly Nguyen and Peter Konstantopolous from the Leukaemia Haemopoiesis Laboratory. Thanks also to Sue Branford and Rebecca Lawrence for their assistance with Quantitative PCR. -

Supplementary Data ASXL2 Regulates Hematopoiesis in Mice and Its
Supplementary data ASXL2 regulates hematopoiesis in mice and its deficiency promotes myeloid expansion Supplementary Methods Genomic DNA extraction Genomic DNA was extracted from BM mononuclear cells using a DNA extraction kit (Puregene Gentra System, Minneapolis, MN, USA) according to the manufacturer’s instructions. Genomic DNA was quantified using Qubit Fluorometer (Life Technologies) and DNA integrity was assessed by agarose gel electrophoresis. For samples with low quantity, DNA was amplified using REPLI-g Ultrafast mini kit (Qiagen). Peripheral blood analysis Complete peripheral blood counts were analysed using Abbott Cell-Dyn 3700 hematology analyzer (Abbott Laboratories). Expression analysis of Asxl2 and Asxl1 Transcript levels of Asxl2 and Asxl1 were estimated using quantitative RT-PCR with following primers: Asxl2 primer set 1, ATTCGACAAGAGATTGAGAAGGAG (forward) and TTTCTGTGAATCTTCAAGGCTTAG (reverse); Asxl2 primer set 2, GCCCTTAACAATGAGTTCTTCACT (forward) and TCCACAGCTCTACTTTCTTCTCCT (reverse); Asxl1 primers, GGTGGAACAATGGAAGGAAA (forward) and CTGGCCGAGAACGTTTCTTA (reverse). Asxl2 protein was detected in immunoblot using anti-ASXL2 antibody (Bethyl). In vitro differentiation of bone marrow cells Bone marrow (BM) cells were cultured in IMDM containing 20% FBS and 10 ng/ml IL3, 10 ng/ml IL6, 20 ng/ml SCF and 20 ng/ml GMCSF for two weeks. For FACS analysis, cells were washed, stained with fluorochrome-conjugated antibodies and analysed on FACS LSR II flow cytometer (BD Biosciences) using FACSDIVA software (BD Biosciences). Colony re-plating assay Bone marrow cells were plated in methylcellulose medium containing mouse stem cell factor (SCF), mouse interleukin 3 (IL-3), human interleukin 6 (IL-6) and human erythropoietin (MethoCult GF M3434; StemCell Technologies). Colonies were enumerated after 9-12 days and cells were harvested for re-plating. -

CD8 Co-Receptor Modifications to Enhance T Cell Immunotherapy
CD8 co-receptor modifications to enhance T cell immunotherapy A thesis submitted to the Division of Infection and Immunity of UCL for the degree of Doctor of Philosophy By Ignatius Chung Chua Department of Immunology Royal Free Hospital Rowland Hill Street London, UK August 2013 0 Declaration Declaration I, IGNATIUS CHUNG CHUA, confirm that this thesis is the result of my own work. Where information has been derived from other sources, I confirm that this has been indicated in the thesis. 1 Abstract Abstract TCR gene transfer can generate tumour antigen-specific T cells for adoptive immunotherapy. Following TCR gene transfer, transduced T cells usually display the same functional avidity as the parental clone from which the TCR was isolated. However, tumour-antigen specific T cells typically recognize over-expressed self- antigen and are often of low/moderate avidity. It is known that optimal recognition of target cells by CTL requires binding of the cognate peptide MHC class I complex (MHCI) by both TCR and the CD8 co-receptor. Some CD8β chain mutations have been shown to increase CD8 binding affinity with peptide/MHCI and enhance T cell effector function. Murine CD8β chain mutants were generated affecting MHC binding sites (L58R, S53L, S54V and L58R/I25A) or glycosylation sites (T120A, T121A, T124A, and T120A/T121A/T124A). The mutated CD8β molecules were introduced into murine splenocytes using retroviral vectors together with tumour antigen-specific TCRs. The CD8β mutants or control CD8β wild type (WT) chains were first introduced into CD8αα T cells obtained from CD8β knockout mice. All T cells were co-transduced to express the murine F5-TCR which recognizes the model tumour antigen, influenza A nucleoprotein (NP366) presented by H2-Db. -

Myeloid Innate Immunity Mouse Vapril2018
Official Symbol Accession Alias / Previous Symbol Official Full Name 2810417H13Rik NM_026515.2 p15(PAF), Pclaf RIKEN cDNA 2810417H13 gene 2900026A02Rik NM_172884.3 Gm449, LOC231620 RIKEN cDNA 2900026A02 gene Abcc8 NM_011510.3 SUR1, Sur, D930031B21Rik ATP-binding cassette, sub-family C (CFTR/MRP), member 8 Acad10 NM_028037.4 2410021P16Rik acyl-Coenzyme A dehydrogenase family, member 10 Acly NM_134037.2 A730098H14Rik ATP citrate lyase Acod1 NM_008392.1 Irg1 aconitate decarboxylase 1 Acot11 NM_025590.4 Thea, 2010309H15Rik, 1110020M10Rik,acyl-CoA Them1, thioesterase BFIT1 11 Acot3 NM_134246.3 PTE-Ia, Pte2a acyl-CoA thioesterase 3 Acox1 NM_015729.2 Acyl-CoA oxidase, AOX, D130055E20Rikacyl-Coenzyme A oxidase 1, palmitoyl Adam19 NM_009616.4 Mltnb a disintegrin and metallopeptidase domain 19 (meltrin beta) Adam8 NM_007403.2 CD156a, MS2, E430039A18Rik, CD156a disintegrin and metallopeptidase domain 8 Adamts1 NM_009621.4 ADAM-TS1, ADAMTS-1, METH-1, METH1a disintegrin-like and metallopeptidase (reprolysin type) with thrombospondin type 1 motif, 1 Adamts12 NM_175501.2 a disintegrin-like and metallopeptidase (reprolysin type) with thrombospondin type 1 motif, 12 Adamts14 NM_001081127.1 Adamts-14, TS14 a disintegrin-like and metallopeptidase (reprolysin type) with thrombospondin type 1 motif, 14 Adamts17 NM_001033877.4 AU023434 a disintegrin-like and metallopeptidase (reprolysin type) with thrombospondin type 1 motif, 17 Adamts2 NM_001277305.1 hPCPNI, ADAM-TS2, a disintegrin and ametalloproteinase disintegrin-like and with metallopeptidase thrombospondin -

Transactivation of Pdgfrb by Dopamine D4 Receptor Does Not
Chi et al. Molecular Brain 2010, 3:22 http://www.molecularbrain.com/content/3/1/22 RESEARCH Open Access Transactivation of PDGFRb by dopamine D4 receptor does not require PDGFRb dimerization Sum Shing Chi1†, Sandra M Vetiska1†, Robin S Gill2, Marilyn S Hsiung2, Fang Liu1,3*, Hubert HM Van Tol1,2,3 Abstract Growth factor-induced receptor dimerization and cross-phosphorylation are hallmarks of signal transduction via receptor tyrosine kinases (RTKs). G protein-coupled receptors (GPCRs) can activate RTKs through a process known as transactivation. The prototypical model of RTK transactivation involves ligand-mediated RTK dimerization and cross-phosphorylation. Here, we show that the platelet-derived growth factor receptor b (PDGFRb) transactivation by the dopamine receptor D4 (DRD4) is not dependent on ligands for PDGFRb. Furthermore, when PDGFRb dimer- ization is inhibited and receptor phosphorylation is suppressed to near basal levels, the receptor maintains its ability to be transactivated and is still effective in signaling to ERK1/2. Hence, the DRD4-PDGFRb-ERK1/2 pathway can occur independently of a PDGF-like ligand, PDGFRb cross-phosphorylation and dimerization, which is distinct from other known forms of transactivation of RTKs by GPCRs. Introduction D2 (DRD2) [5-7], b2 adrenergic receptor [8], M1 mus- Receptor tyrosine kinases (RTKs) consist of a large carinic receptor [9], angiotensin II receptor [10], lyso- family of receptors whose members serve a wide range phosphatidic acid (LPA) receptor [11], ET1 receptor of physiological functions including growth, differentia- [12] and thrombin receptor [12] have been shown to tion and synaptic modulation. The members of this transactivate either the epidermal growth factor receptor receptor family generally feature an extracellular ligand- (EGFR) or the platelet-derived growth factor receptor b binding domain, linked by a transmembrane domain to (PDGFRb). -

B Cell Receptor Signaling in Receptor Editing and Leukemia
B CELL RECEPTOR SIGNALING IN RECEPTOR EDITING AND LEUKEMIA A DISSERTATION SUBMITTED TO THE FACULTY OF THE GRADUATE SCHOOL OF THE UNIVERSITY OF MINNESOTA BY LAURA BAILEY RAMSEY IN PARTIAL FULFILLMENT OF THE REQUIREMENTS FOR THE DEGREE OF DOCTOR OF PHILOSOPHY MICHAEL A. FARRAR, ADVISER JULY 2009 © Laura Bailey Ramsey 2009 i Acknowledgements I would like to thank my family and friends for all their support, for without it I would not have accomplished all that I have. I would especially like to thank my husband Colin, for supporting me through the tough times and celebrating with me during the happy times. I am eternally grateful to my parents and grandparents for their support and unconditional love. These last five years would not have been the same without the support of my “Minnesota Mom,” Lyn Glenn. They have all encouraged me to accomplish my dreams, and without them I would not have been able to do it. I would like to thank the Behrens lab for all their support and patience with me when I started graduate school. They introduced me to the world of immunology and B cells, and for that I am forever grateful. Tim Behrens was a great mentor and project director for the first two years of my graduate career. Brian Schram and Keli Hippen were especially helpful in teaching me all about B cells. Jason Bauer, Thearith Koeuth, and Emily Gillespie have taught me many things about microarrays and continue to support me at the University of Minnesota. Jessica Oehrlein and Amanda Vegoe took wonderful care of my mice. -

Vitro Generated to Ex Vivo CD8 T Cells Comparative and Functional
Downloaded from http://www.jimmunol.org/ by guest on September 27, 2021 is online at: average * The Journal of Immunology , 21 of which you can access for free at: 2012; 189:3411-3420; Prepublished online 27 from submission to initial decision 4 weeks from acceptance to publication Comparative and Functional Evaluation of In Vitro Generated to Ex Vivo CD8 T Cells Dzana D. Dervovic, Maria Ciofani, Korosh Kianizad and Juan Carlos Zúñiga-Pflücker J Immunol August 2012; doi: 10.4049/jimmunol.1200979 http://www.jimmunol.org/content/189/7/3411 cites 61 articles Submit online. Every submission reviewed by practicing scientists ? is published twice each month by Receive free email-alerts when new articles cite this article. Sign up at: http://jimmunol.org/alerts http://jimmunol.org/subscription Submit copyright permission requests at: http://www.aai.org/About/Publications/JI/copyright.html http://www.jimmunol.org/content/suppl/2012/08/28/jimmunol.120097 9.DC1 This article http://www.jimmunol.org/content/189/7/3411.full#ref-list-1 Information about subscribing to The JI No Triage! Fast Publication! Rapid Reviews! 30 days* Why • • • Material References Permissions Email Alerts Subscription Supplementary The Journal of Immunology The American Association of Immunologists, Inc., 1451 Rockville Pike, Suite 650, Rockville, MD 20852 Copyright © 2012 by The American Association of Immunologists, Inc. All rights reserved. Print ISSN: 0022-1767 Online ISSN: 1550-6606. This information is current as of September 27, 2021. The Journal of Immunology Comparative and Functional Evaluation of In Vitro Generated to Ex Vivo CD8 T Cells Dzˇana D. Dervovic´,*,† Maria Ciofani,*,†,1 Korosh Kianizad,*,† and Juan Carlos Zu´n˜iga-Pflu¨cker*,† The generation of the cytotoxic CD8 T cell response is dependent on the functional outcomes imposed by the intrathymic constraints of differentiation and self-tolerance. -
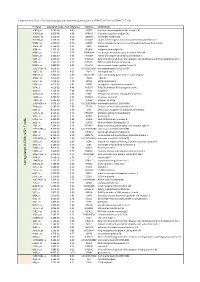
Up Regu Lated in C D 8+ILT2+ T Cells
Supplementary Table I. Specifically upregulated and downregulated genes in CD8+ILT2+ T cells vs CD8+ILT2- T cells Probeset adjusted P Value Fold difference SYMBOL GENENAME 10859_at 2.09E-04 8.81 LILRB1 leukocyte immunoglobulin like receptor B1 353345_at 9.81E-04 6.69 GPR141 G protein-coupled receptor 141 162966_at 1.25E-03 6.29 ZNF600 zinc finger protein 600 353189_at 1.63E-03 5.79 SLCO4C1 solute carrier organic anion transporter family member 4C1 59084_at 1.63E-03 5.76 ENPP5 ectonucleotide pyrophosphatase/phosphodiesterase 5 (putative) 10578_at 1.75E-03 5.64 GNLY granulysin 3684_at 1.75E-03 5.64 ITGAM integrin subunit alpha M 64097_at 2.13E-03 5.33 EPB41L4A erythrocyte membrane protein band 4.1 like 4A 81553_at 2.13E-03 5.35 FAM49A family with sequence similarity 49 member A 3804_at 2.24E-03 5.24 KIR2DL3 killer cell immunoglobulin like receptor, two Ig domains and long cytoplasmic tail 3 9788_at 2.36E-03 5.15 MTSS1 MTSS1, I-BAR domain containing 81563_at 2.99E-03 4.93 C1orf21 chromosome 1 open reading frame 21 102724104_at 3.61E-03 4.74 LOC102724104 uncharacterized LOC102724104 10875_at 3.91E-03 4.65 FGL2 fibrinogen like 2 284367_at 3.94E-03 4.60 SIGLEC17P sialic acid binding Ig like lectin 17, pseudogene 30009_at 3.97E-03 4.57 TBX21 T-box 21 23314_at 4.01E-03 4.48 SATB2 SATB homeobox 2 53637_at 4.01E-03 4.45 S1PR5 sphingosine-1-phosphate receptor 5 5874_at 4.01E-03 4.46 RAB27B RAB27B, member RAS oncogene family 4646_at 4.75E-03 4.16 MYO6 myosin VI 445347_at 5.02E-03 4.09 TARP TCR gamma alternate reading frame protein 11098_at 5.62E-03 -

Antibodies Against T Cell Immunoglobulin Domain And
(19) TZZ__¥Z_T (11) EP 1 613 750 B1 (12) EUROPEAN PATENT SPECIFICATION (45) Date of publication and mention (51) Int Cl.: of the grant of the patent: C12N 15/12 (2006.01) C07K 14/47 (2006.01) 14.10.2015 Bulletin 2015/42 C07K 16/18 (2006.01) A61P 35/00 (2006.01) (21) Application number: 04757910.7 (86) International application number: PCT/US2004/008502 (22) Date of filing: 19.03.2004 (87) International publication number: WO 2004/084823 (07.10.2004 Gazette 2004/41) (54) ANTIBODIES AGAINST T CELL IMMUNOGLOBULIN DOMAIN AND MUCIN DOMAIN 1 (TIM-1) ANTIGEN AND USES THEREOF ANTIKÖRPER GEGEN DAS T- ZELLEN-IMMUNGLOBULINDOMÄNE- UND MUCINDOMÄNE- 1 (TIM- 1) ANTIGEN UND VERWENDUNGEN DAVON ANTICORPS CONTRE L’ANTIGENE DE LYMPHOCYTES T, DU DOMAINE D’IMMUNOGLOBULINE ET DU DOMAINE 1 DE MUCINE (TIM-1) ET LEURS UTILISATIONS (84) Designated Contracting States: • MESRI, Mehdi AT BE BG CH CY CZ DE DK EE ES FI FR GB GR North Potomac, MD 20878 (US) HU IE IT LI LU MC NL PL PT RO SE SI SK TR (74) Representative: Giles, Ashley Simon et al (30) Priority: 19.03.2003 US 456652 P Haseltine Lake LLP Lincoln House, 5th Floor (43) Date of publication of application: 300 High Holborn 11.01.2006 Bulletin 2006/02 London WC1V 7JH (GB) (73) Proprietor: Amgen Fremont Inc. (56) References cited: Fremont, CA 94555 (US) WO-A-02/098920 WO-A-03/002722 WO-A-03/080856 WO-A-2005/001092 (72) Inventors: WO-A1-97/44460 US-B1- 6 664 385 • LANDES, Gregory, M.