Structures and Antifungal Activities in Foods by Nuanyi Liang a Thesis
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

(12) United States Patent (10) Patent No.: US 6,692,728 B2 Weipert Et Al
USOO6692728B2 (12) United States Patent (10) Patent No.: US 6,692,728 B2 Weipert et al. (45) Date of Patent: Feb. 17, 2004 (54) POLYESTERS BASED ON HYDROXY FATTY (52) U.S. Cl. ......................... 424/59; 424/497; 424/489; ACDS AND LOWER HYDROXY ALKYL 424/70.11; 424/78.37; 424/78.08; 514/785; ACDS AND USES THEREOF 560/171; 560/172; 560/176; 560/183; 525/400 (58) Field of Search .......................... 424/59, 497, 489, (75) Inventors: Paul David Weipert, High Point, NC 424/70.11, 78.37; 514/785; 560/172,176, (US); Bharat B. Desai, Spartanburg, 183; 525/400 SC (US) (73) Assignee: Ethox Chemicals LLC, Greenville, SC ") References Cited (US) U.S. PATENT DOCUMENTS ( c: ) Notice: Subject to any disclaimer, the term of this 5,502,116 A 3/1996 Noda ......................... 525/415 patent is extended or adjusted under 35 5,614,576 A * 3/1997 Rutherford et al. ......... 524/270 U.S.C. 154(b) by 0 days. 5,851,937 A * 12/1998 Wu et al. ................... 442/394 * cited by examiner (21) Appl. No.: 10/388,426 (22) Filed: Mar 17, 2003 Primary Examiner Sabiha Qazi 9 (74) Attorney, Agent, or Firm-Isaac A. Angres (65) Prior Publication Data (57) ABSTRACT US 2003/0175222 A1 Sep. 18, 2003 The present invention provides biodegradable polyesters Related U.S. Application Data based on lower hydroxy acids and hydroxy fatty acids. The resulting polyesters are useful as cosmetic vehicles for (62) Division of application No. 09/805,894, filed on Mar. 15, Sunscreens, skin lotions and by themselves are also useful as 2001, now Pat. -

Assessment Report on Ricinus Communis L., Oleum Final
2 February 2016 EMA/HMPC/572973/2014 Committee on Herbal Medicinal Products (HMPC) Assessment report on Ricinus communis L., oleum Final Based on Article 10a of Directive 2001/83/EC as amended (well-established use) Herbal substance(s) (binomial scientific name Ricinus communis L., oleum (castor oil) of the plant, including plant part) Herbal preparation Fatty oil obtained from seeds of Ricinus communis L. by cold expression Pharmaceutical forms Herbal preparation in liquid or solid dosage forms for oral use Rapporteur C. Purdel Peer-reviewer B. Kroes 30 Churchill Place ● Canary Wharf ● London E14 5EU ● United Kingdom Telephone +44 (0)20 3660 6000 Facsimile +44 (0)20 3660 5555 Send a question via our website www.ema.europa.eu/contact An agency of the European Union © European Medicines Agency, 2016. Reproduction is authorised provided the source is acknowledged. Table of contents Table of contents ................................................................................................................... 2 1. Introduction ....................................................................................................................... 4 1.1. Description of the herbal substance(s), herbal preparation(s) or combinations thereof .. 4 1.2. Search and assessment methodology ..................................................................... 6 2. Data on medicinal use ........................................................................................................ 6 2.1. Information about products on the market ............................................................. -

Castor Oil Induces Laxation and Uterus Contraction Via Ricinoleic Acid Activating Prostaglandin EP3 Receptors Sorin Tunarua, Till F
Castor oil induces laxation and uterus contraction via ricinoleic acid activating prostaglandin EP3 receptors Sorin Tunarua, Till F. Althoffa, Rolf M. Nüsingb, Martin Dienerc, and Stefan Offermannsa,d,1 aDepartment of Pharmacology, Max-Planck-Institute for Heart and Lung Research, 61231 Bad Nauheim, Germany; dMedical Faculty, bInstitute for Clinical Pharmacology, J. W. Goethe University Frankfurt, 60590 Frankfurt, Germany; and cInstitute for Veterinary Physiology, Justus Liebig University, 35392 Giessen, Germany Edited by John H. Exton, Vanderbilt University School of Medicine, Nashville, TN, and approved April 25, 2012 (received for review January 30, 2012) Castor oil is one of the oldest drugs. When given orally, it has by the enteric nervous system or are direct effects on intestinal a laxative effect and induces labor in pregnant females. The smooth muscle remained unclear. effects of castor oil are mediated by ricinoleic acid, a hydroxylated The present study was undertaken to elucidate the molecular fatty acid released from castor oil by intestinal lipases. Despite the mechanism underlying the biological effect of castor oil-derived wide-spread use of castor oil in conventional and folk medicine, ricinoleic acid. Based on cellular signaling studies and an siRNA the molecular mechanism by which ricinoleic acid acts remains screening approach, we identified prostaglandin E2 receptors as unknown. Here we show that the EP3 prostanoid receptor is spe- targets of ricinoleic acid and show that the EP3 receptor medi- cifically activated by ricinoleic acid and that it mediates the phar- ates the effects of castor oil on the motility of the uterus and macological effects of castor oil. In mice lacking EP3 receptors, the the intestine. -
Poster Presenter Groups
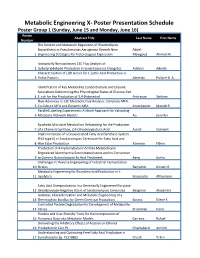
Metabolic Engineering X- Poster Presentation Schedule Poster Group 1 (Sunday, June 15 and Monday, June 16) Poster Abstract Title Last Name First Name Number The Genetic and Metabolic Regulation of Rhamnolipids Biosynthesis in Pseudomonas Aeruginosa Reveals New Abdel- 1 Engineering Strategies for Heterologous Expression Mawgoud Ahmad M. Isotopically Nonstationary 13C Flux Analysis of 2 Isobutyraldehyde Production in Synechococcus Elongatus Adebiyi Adeola Characterization of LDH Genes for L-Lactic Acid Production in 3 Pichia Pastoris Almeida Pollyne B. A. Identification of Key Metabolite Concentrations and Enzyme Saturations Determining the Physiological States of Glucose-Fed 4 E. coli for the Production of 1,4-Butanediol Andreozzi Stefano New Advances in 13C Metabolic Flux Analysis: Complete-MFA, 5 Co-Culture MFA and Dynamic MFA Antoniewicz Maciek R. Parallel Labeling Experiments: A Novel Approach for Validating 6 Metabolic Network Models Au Jennifer Synthetic Microbial Metabolism Refactoring for the Production 7 of a Chemical Synthon, 2,4-Dihydroxybutyric Acid Auriol Clément Implementation of a Disassociated Fatty Acid Synthase System (FAS type II) in Saccharomyces Cerevisiae for Fatty Acid and 8 Wax Ester Production Azevedo Flávio Production of 4-Hydroxybutyric Acid By Metabolically Engineered Mannheimia Succiniciproducens and Its Conversion 9 to Gamma-Butyrolactone By Acid Treatment Bang Junho Challenges in Reverse Engineering of Industrial Fermentation 10 Strains Benjamin Kirsten R. Metabolic Engineering for Ricinoleic Acid Production in Y. 11 Lipolytica Beopoulos Athanasios Fatty Acid Overproduction in a Genetically Engineered Puryvate 12 Decarboxylase-Negative Strain of Saccharomyces Cerevisiae Bergman Alexandra Isolation, Characterization and Metabolic Engineering of a 13 Thermophilic Bacillus for Green Chemical Production Bosma Elleke F. -

Dr. Duke's Phytochemical and Ethnobotanical Databases List of Chemicals for Dysmenorrhea
Dr. Duke's Phytochemical and Ethnobotanical Databases List of Chemicals for Dysmenorrhea Chemical Activity Count (+)-ADLUMINE 1 (+)-ALLOMATRINE 1 (+)-ALPHA-VINIFERIN 1 (+)-BORNYL-ISOVALERATE 1 (+)-CATECHIN 4 (+)-EUDESMA-4(14),7(11)-DIENE-3-ONE 1 (+)-GALLOCATECHIN 1 (+)-HERNANDEZINE 1 (+)-ISOCORYDINE 2 (+)-PSEUDOEPHEDRINE 1 (+)-T-CADINOL 1 (-)-16,17-DIHYDROXY-16BETA-KAURAN-19-OIC 1 (-)-ALPHA-BISABOLOL 3 (-)-ANABASINE 1 (-)-ARGEMONINE 1 (-)-BETONICINE 1 (-)-BORNYL-CAFFEATE 1 (-)-BORNYL-FERULATE 1 (-)-BORNYL-P-COUMARATE 1 (-)-DICENTRINE 2 (-)-EPIAFZELECHIN 1 (-)-EPICATECHIN 1 (-)-EPIGALLOCATECHIN-GALLATE 1 (1'S)-1'-ACETOXYCHAVICOL-ACETATE 1 (15:1)-CARDANOL 1 (E)-4-(3',4'-DIMETHOXYPHENYL)-BUT-3-EN-OL 1 1,7-BIS-(4-HYDROXYPHENYL)-1,4,6-HEPTATRIEN-3-ONE 1 Chemical Activity Count 1,8-CINEOLE 6 10-ACETOXY-8-HYDROXY-9-ISOBUTYLOXY-6-METHOXYTHYMOL 1 10-DEHYDROGINGERDIONE 1 10-GINGERDIONE 1 11-HYDROXY-DELTA-8-THC 1 11-HYDROXY-DELTA-9-THC 1 12,118-BINARINGIN 1 12-ACETYLDEHYDROLUCICULINE 1 13',II8-BIAPIGENIN 1 13-OXYINGENOL-ESTER 1 16,17-DIHYDROXY-16BETA-KAURAN-19-OIC 1 16-EPIMETHUENINE 1 16-HYDROXYINGENOL-ESTER 1 2'-HYDROXY-FLAVONE 1 2'-O-GLYCOSYLVITEXIN 1 2-BETA,3BETA-27-TRIHYDROXYOLEAN-12-ENE-23,28-DICARBOXYLIC-ACID 1 2-METHYLBUT-3-ENE-2-OL 1 20-DEOXYINGENOL-ESTER 1 22BETA-ESCIN 1 24-METHYLENE-CYCLOARTANOL 2 3,4-DIMETHOXYTOLUENE 1 3,4-METHYLENE-DIOXYCINNAMIC-ACID-BORNYL-ESTER 2 3,4-SECOTRITERPENE-ACID-20-EPI-KOETJAPIC-ACID 1 3-ACETYLACONITINE 3 3-ACETYLNERBOWDINE 1 3-BETA-ACETOXY-20,25-EPOXYDAMMARANE-24-OL 1 3-BETA-HYDROXY-2,3-DIHYDROWITHANOLIDE-F -

Dr. Duke's Phytochemical and Ethnobotanical Databases List of Chemicals for Epilepsy
Dr. Duke's Phytochemical and Ethnobotanical Databases List of Chemicals for Epilepsy Chemical Activity Count (+)-ALPHA-VINIFERIN 1 (+)-BORNYL-ISOVALERATE 1 (+)-CATECHIN 3 (+)-EUDESMA-4(14),7(11)-DIENE-3-ONE 1 (+)-HERNANDEZINE 1 (+)-ISOCORYDINE 1 (+)-PSEUDOEPHEDRINE 1 (+)-SYRINGARESINOL-DI-O-BETA-D-GLUCOSIDE 1 (+)-T-CADINOL 1 (-)-16,17-DIHYDROXY-16BETA-KAURAN-19-OIC 1 (-)-ALPHA-BISABOLOL 2 (-)-ANABASINE 1 (-)-APOGLAZIOVINE 1 (-)-BETONICINE 1 (-)-BORNYL-CAFFEATE 1 (-)-BORNYL-FERULATE 1 (-)-BORNYL-P-COUMARATE 1 (-)-DICENTRINE 2 (-)-EPIAFZELECHIN 1 (-)-EPICATECHIN 1 (-)-EPIGALLOCATECHIN-GALLATE 1 (1'S)-1'-ACETOXYCHAVICOL-ACETATE 1 (15:1)-CARDANOL 1 (E)-4-(3',4'-DIMETHOXYPHENYL)-BUT-3-EN-OL 1 1,7-BIS-(4-HYDROXYPHENYL)-1,4,6-HEPTATRIEN-3-ONE 1 1,8-CINEOLE 4 10-ACETOXY-8-HYDROXY-9-ISOBUTYLOXY-6-METHOXYTHYMOL 1 Chemical Activity Count 10-DEHYDROGINGERDIONE 1 10-GINGERDIONE 1 11-HYDROXY-DELTA-8-THC 1 11-HYDROXY-DELTA-9-THC 1 13',II8-BIAPIGENIN 1 13-OXYINGENOL-ESTER 1 16,17-DIHYDROXY-16BETA-KAURAN-19-OIC 1 16-EPIMETHUENINE 1 16-HYDROXYINGENOL-ESTER 1 2'-O-GLYCOSYLVITEXIN 1 2-BETA,3BETA-27-TRIHYDROXYOLEAN-12-ENE-23,28-DICARBOXYLIC-ACID 1 2-METHYLBUT-3-ENE-2-OL 2 20-DEOXYINGENOL-ESTER 1 22BETA-ESCIN 1 24-METHYLENE-CYCLOARTANOL 1 3,3'-DIMETHYLELLAGIC-ACID 1 3,4-DIMETHOXYTOLUENE 1 3,4-METHYLENE-DIOXYCINNAMIC-ACID-BORNYL-ESTER 2 3,4-SECOTRITERPENE-ACID-20-EPI-KOETJAPIC-ACID 1 3-ACETYLACONITINE 1 3-ACETYLNERBOWDINE 1 3-BETA-HYDROXY-2,3-DIHYDROWITHANOLIDE-F 1 3-HYDROXY-FLAVONE 1 3-N-BUTYL-PHTHALIDE 3 3-O-ACETYLOLEANOLIC-ACID 1 3-OXO-11-ALPHA-HYDROXYOLEAN-12-ENE-30-OIC-ACID -

Effects of Oleic and Ricinoleic Acids on Net Jejunal Water and Electrolyte Movement
Effects of Oleic and Ricinoleic Acids on Net Jejunal Water and Electrolyte Movement PERFUSION STUDIES IN MAN HELMUT V. AMMON, PAUL J. THOMAS, and SIDNEY F. PHILLIPS From the Gastroenterology Unit, Mayo Clinic and Mayo Foundation, Rochester, Minnesota 55901 ABSTRA CT To examine the effects of oleic acid by bacterial hydration of unsaturated dietary fatty acids and ricinoleic acid on jejunal absorption, steady-state (2), are chemically similar to a known cathartic, ricino- jejunal perfusions were performed in healthy volunteers. leic acid. This hypothesis of the action of hydroxy fatty Taurocholate, used to solubilize the fatty acids, did not acids is supported by the results of other investigations: influence absorption. Both fatty acids (concentration, the study of animals with an experimental "blind loop 10 mM) reversed electrolyte and water net movement; syndrome" (3), perfusion of the intestine with fatty that is, they induced fluid secretion; this effect was acid solutions (4), and analysis of steatorrheal stools rapidly reversible. Ricinoleic acid (the active principle in man (5). Specifically, hydroxy fatty acids provoke of castor oil) was the more potent, producing fluid se- water secretion in the rat colon (4) and impair net cretion when perfused at concentrations at which oleic fluid movement in the human colon (6). In the canine acid was without effect. However, ricinoleic acid was ileum, however, water absorption has been blocked not absorbed more slowly than was oleic acid, and hence only by 10-hydroxystearic acid and ricinoleic acid (the was associated with higher intraluminal concentrations. active principle of castor oil) but also by oleic acid (7). -

Tepzz Z89z ¥B T
(19) TZZ ZZ_¥_T (11) EP 2 089 013 B1 (12) EUROPEAN PATENT SPECIFICATION (45) Date of publication and mention (51) Int Cl.: of the grant of the patent: A61K 31/167 (2006.01) A61K 31/192 (2006.01) 27.09.2017 Bulletin 2017/39 A61K 45/06 (2006.01) A61P 29/00 (2006.01) (21) Application number: 07854106.7 (86) International application number: PCT/US2007/081589 (22) Date of filing: 17.10.2007 (87) International publication number: WO 2008/051759 (02.05.2008 Gazette 2008/18) (54) COMBINATION OF ACETAMINOPHEN WITH IBUPROFEN FOR THE TREAMENT OF PAIN KOMBINATION VON ACETAMINOPHEN MIT IBUPROFEN ZUR BEHANDLUNG VON SCHMERZEN. COMBINAISON DE ACÉTAMINOPHÈNE ET IBUPROFÈNE POUR LE TRAITEMENT DE DOULEUR (84) Designated Contracting States: (74) Representative: Kirsch, Susan Edith et al AT BE BG CH CY CZ DE DK EE ES FI FR GB GR Carpmaels & Ransford LLP HU IE IS IT LI LT LU LV MC MT NL PL PT RO SE One Southampton Row SI SK TR London WC1B 5HA (GB) (30) Priority: 20.10.2006 US 853509 P (56) References cited: EP-A- 0 012 621 EP-A- 0 109 281 (43) Date of publication of application: WO-A-2006/004449 US-A- 5 409 709 19.08.2009 Bulletin 2009/34 • MIRANDA ET AL: "Synergism between (73) Proprietor: Johnson & Johnson Consumer Inc. paracetamol and nonsteroidal anti-inflammatory Skillman, NJ 08558 (US) drugs in experimental acute pain" PAIN, ELSEVIER SCIENCE PUBLISHERS, (72) Inventors: AMSTERDAM, NL, vol. 121, no. 1-2, March 2006 • SWANN, Jim (2006-03), pages 22-28, XP005330893 ISSN: Waterloo, ON N2L 5Y8 (CA) 0304-3959 • CUMMINS, Peter Guelph, ON N1L 1B4 (CA) Note: Within nine months of the publication of the mention of the grant of the European patent in the European Patent Bulletin, any person may give notice to the European Patent Office of opposition to that patent, in accordance with the Implementing Regulations. -

Assessment Report on Ricinus Communis L., Oleum Draft
7 July 2015 EMA/HMPC/572973/2014 Committee on Herbal Medicinal Products (HMPC) Assessment report on Ricinus communis L., oleum Draft Based on Article 10a of Directive 2001/83/EC as amended (well-established use) Herbal substance(s) (binomial scientific name of the plant, including plant part) Ricinus communis L., oleum (castor oil) Herbal preparation Fatty oil obtained from seeds of Ricinus communis L. by cold expression Pharmaceutical forms Herbal preparation in liquid or solid dosage forms for oral use Rapporteur C. Purdel Peer-reviewer B. Kroes Note: This draft assessment report is published to support the public consultation of the draft European Union herbal monograph on Ricinus communis L., oleum. It is a working document, not yet edited, and shall be further developed after the release for consultation of the monograph. Interested parties are welcome to submit comments to the HMPC secretariat, which will be taken into consideration but no ‘overview of comments received during the public consultation’ will be prepared on comments that will be received on this assessment report. The publication of this draft assessment report has been agreed to facilitate the understanding by Interested Parties of the assessment that has been carried out so far and led to the preparation of the draft monograph. 30 Churchill Place ● Canary Wharf ● London E14 5EU ● United Kingdom Telephone +44 (0)20 3660 6000 Facsimile +44 (0)20 3660 5555 Send a question via our website www.ema.europa.eu/ contact An agency of the European Union © European Medicines Agency, 2015. Reproduction is authorised provided the source is acknowledged. Table of contents Table of contents .................................................................................................................. -

Evaluation of the Anti-Diarrheal Activity of the Leaf Extract of Croton Macrostachyus Hocsht. Ex Del.(Euphorbiaceae) in Mice Model
Degu et al. BMC Complementary and Alternative Medicine (2016) 16:379 DOI 10.1186/s12906-016-1357-9 RESEARCH ARTICLE Open Access Evaluation of the anti-diarrheal activity of the leaf extract of Croton macrostachyus Hocsht. ex Del. (Euphorbiaceae) in mice model Amsalu Degu1* , Ephrem Engidawork2 and Workineh Shibeshi2 Abstract Background: Traditional healers in Ethiopia use a wide range of medicinal plants with antidiarrheal properties. Among these, Croton macrostachyus is one such plant claimed to have an antidiarrheal activity in Ethiopian folklore medicine. Previous studies showed that the crude extract is endowed with the claimed property. The present study was undertaken to further the claim by screening different fractions for the said activity so that it could serve as a basis for subsequent studies. Methods: The fractions were obtained by successive extraction in soxhlet apparatus with solvents of different polarity (chloroform & methanol) followed by cold maceration of the deposit of the methanol fraction with distilled water. The antidiarrheal activity was evaluated using castor oil induced diarrheal model, charcoal meal test and anti-enteropooling test in mice. The test groups received various doses (300, 400, 500 mg/kg and an additional dose of 1000 mg/kg for the aqueous fraction) of the fractions, whereas positive controls received either Loperamide (3 mg/kg) or Atropine (5 mg/kg) and negative controls received vehicle (10 ml/kg). Results: In the castor oil induced model, the chloroform (at all test doses) and methanol (at 400 & 500 mg/kg) fractions significantly delayed diarrheal onset, decreased stool frequency and weight of feces. The aqueous fraction was however devoid of significant effect at all the tested doses. -

Faecal Free Fatty Acids in Tropical Sprue and Their Inhibition of Atpases
Gut: first published as 10.1136/gut.24.4.300 on 1 April 1983. Downloaded from Gut, 1983. 24, 30W-305 Faecal free fatty acids in tropical sprue and their possible role in the production of diarrhoea by inhibition of ATPases C TIRUPPATHI, K A BALASUBRAMANIAN, P G HILL, AND V I MATHAN From the Wellcome Research Unit, Christian Medical College Hospital, Vellore, India SUMMARY Faecal excretion of fatty acids is increased in patients with tropical sprue because of unabsorbed dietary fatty acids. The excretion of fatty acids correlates well with faecal wet weight. In vitro unsaturated fatty acids inhibited Na K-ATPase and Mg-ATPase isolated from basolateral membranes of enterocytes and colonocytes. These findings are a possible explanation for the observed abnormalities in water and electrolyte absorption by the colon in patients with tropical sprue and steatorrhoea. Colonic water and electrolyte absorption is defective ward diet containing 50 g fat per day and the analysis in patients with tropical sprue.' Faecal free fatty was done after at least one week on this diet. The acids could play a role in the pathogenesis of this dietary fat was mnainly groundnut oil and analysis of functional defect of the colon in patients with a typical 24 hour diet showed that palmitic (16:0), tropical sprue and steatorrhoea. The precise role of oleic (18: 1). and linoleic (18:2) acids were the major http://gut.bmj.com/ fatty acids in the genesis of diarrhoea associated fatty acids (Fig. 1). with steatorrhoea is not clear' but it has been Faeces were passed into polyethylene containers suggested that hydroxv fatty acids could produce a and kept immediately by the patients in a freezer secretory diarrhoea.3 This paper reports the (-200C). -

Diacylglycerol Acyltransferase Activity and Triacylglycerol Synthesis in Germinating Castor Seed Cotyledons Xiaohua He, Grace Q
Diacylglycerol Acyltransferase Activity and Triacylglycerol Synthesis in Germinating Castor Seed Cotyledons Xiaohua He, Grace Q. Chen, Jiann-Tsyh Lin, and Thomas A. McKeon* Western Regional Research Center, USDA, Albany, California 94710 ABSTRACT: The central importance of storage lipid break- erolipid synthesis were present and active in soybean cotyle- down in providing carbon and energy during seed germination dons during seed germination (4). The synthesis of TAG has has been demonstrated by isolating the genes encoding the en- also been shown to occur during seed germination of some zymes involved in FA β-oxidation. In contrast, little is known plants, such as the pea (5), cucumber (6), and soybean (7). about the ability of germinating seeds to synthesize TAG. We These observations suggest the presence of DAG acyltrans- report that castor cotyledons are capable of TAG synthesis. The ferase (DGAT) in germinating seeds. Recently, we identified a rate of incorporation of ricinoleic acid into TAG reached a peak cDNA encoding DGAT from castor seed (RcDGAT) based on at 7 d after imbibition (DAI) (1.14 nmol/h/mg) and decreased rapidly thereafter, but was sustained at 20 DAI in cotyledons its homology to other plant-type DGAT1 cDNA (8) and inves- and true leaves. The castor DAG acyltransferase (RcDGAT) tigated the expression of the RcDGAT gene and DGAT activ- mRNA and protein were expressed throughout seed germina- ity in developing seeds (9). In this study we extend our exami- tion at levels considerably enhanced from that in the dormant nation of the RcDGAT gene and protein to cotyledons of ger- seed, thus indicating new expression.