The Natural Radioactive Isotopes of Beryllium in the Environment
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

The Radiochemistry of Beryllium
National Academy of Sciences National Research Council I NUCLEAR SCIENCE SERIES The Radiochemistry ·of Beryllium COMMITTEE ON NUCLEAR SCIENCE L. F. CURTISS, Chairman ROBLEY D. EVANS, Vice Chairman National Bureau of Standards MassaChusetts Institute of Technol0gy J. A. DeJUREN, Secretary ./Westinghouse Electric Corporation H.J. CURTIS G. G. MANOV Brookhaven National' LaboratOry Tracerlab, Inc. SAMUEL EPSTEIN W. WAYNE MEINKE CalUornia Institute of Technology University of Michigan HERBERT GOLDSTEIN A.H. SNELL Nuclear Development Corporation of , oak Ridge National Laboratory America E. A. UEHLING H.J. GOMBERG University of Washington University of Michigan D. M. VAN PATTER E.D.KLEMA Bartol Research Foundation Northwestern University ROBERT L. PLATZMAN Argonne National Laboratory LIAISON MEMBERS PAUL C .. AEBERSOLD W.D.URRY Atomic Energy Commission U. S. Air Force J. HOW ARD McMILLEN WILLIAM E. WRIGHT National Science Foundation Office of Naval Research SUBCOMMITTEE ON RADIOCHEMISTRY W. WAYNE MEINKE, Chairman HAROLD KIRBY University of Michigan Mound Laboratory GREGORY R. CHOPPIN GEORGE LEDDICOTTE Florida State University. Oak Ridge National Laboratory GEORGE A. COW AN JULIAN NIELSEN Los Alamos Scientific Laboratory Hanford Laboratories ARTHUR W. FAIRHALL ELLIS P. STEINBERG University of Washington Argonne National Laboratory JEROME HUDIS PETER C. STEVENSON Brookhaven National Laboratory University of California (Livermore) EARL HYDE LEO YAFFE University of CalUornia (Berkeley) McGill University CONSULTANTS NATHAN BALLOU WILLIAM MARLOW Naval Radiological Defense Laboratory N atlonal Bureau of Standards JAMESDeVOE University of Michigan CHF.MISTRY-RADIATION AND RADK>CHEMIST The Radiochemistry of Beryllium By A. W. FAIRHALL. Department of Chemistry University of Washington Seattle, Washington May 1960 ' Subcommittee on Radiochemistry National Academy of Sciences - National Research Council Printed in USA. -

Measurement of 10Be from Lake Malawi (Africa) Drill Core Sediments and Implications for Geochronology
Palaeogeography, Palaeoclimatology, Palaeoecology 303 (2011) 110–119 Contents lists available at ScienceDirect Palaeogeography, Palaeoclimatology, Palaeoecology journal homepage: www.elsevier.com/locate/palaeo Measurement of 10Be from Lake Malawi (Africa) drill core sediments and implications for geochronology L.R. McHargue a,⁎, A.J.T. Jull b, A. Cohen a a Department of Geosciences, University of Arizona, 1040 E. Fourth Street, Tucson, Arizona 85721, United States b NSF Arizona AMS Laboratory, University of Arizona, 1118 E. Fourth Street, Tucson, Arizona 85721, United States article info abstract Article history: The cosmogenic radionuclide 10Be was measured from drill core sediments from Lake Malawi in order to Received 19 August 2008 help construct a chronology for the study of the tropical paleoclimate in East Africa. Sediment samples were Received in revised form 29 January 2010 taken every 10 m from the core MAL05-1C to 80 m in depth and then from that depth in core MAL05-1B to Accepted 4 February 2010 382 m. Sediment samples were then later taken at a higher resolution of every 2 m from MAL05-1C. They Available online 12 February 2010 were then leached to remove the authigenic fraction, the leachate was processed to separate out the beryllium isotopes, and 10Be was measured at the TAMS Facility at the University of Arizona. The 10Be/9Be Keywords: fi Beryllium-10 pro le from Lake Malawi sediments is similar to those derived from marine sediment cores for the late Cosmogenic radionuclide Pleistocene, and is consistent with the few radiocarbon and OSL IR measurements made from the same core. Lake sediments Nevertheless, a strong correlation between the stable isotope 9Be and the cosmogenic isotope 10Be suggests Lake Malawi that both isotopes have been well mixed before deposition unlike in some marine sediment cores. -

Meteoric Be and Be As Process Tracers in the Environment
Chapter 5 Meteoric 7Be and 10Be as Process Tracers in the Environment James M. Kaste and Mark Baskaran 7 10 Abstract Be (T1/2 ¼ 53 days) and Be (T1/2 ¼ occurring Be isotopes of use to Earth scientists are the 7 1.4 Ma) form via natural cosmogenic reactions in the short-lived Be (T1/2 ¼ 53.1 days) and the longer- 10 atmosphere and are delivered to Earth’s surface by wet lived Be (T1/2 ¼ 1.4 Ma; Nishiizumi et al. 2007). and dry deposition. The distinct source term and near- Because cosmic rays that cause the initial cascade of constant fallout of these radionuclides onto soils, vege- neutrons and protons in the upper atmosphere respon- tation, waters, ice, and sediments makes them valuable sible for the spallation reactions are attenuated by tracers of a wide range of environmental processes the mass of the atmosphere itself, production rates of operating over timescales from weeks to millions of comsogenic Be are three orders of magnitude higher in years. Beryllium tends to form strong bonds with oxygen the stratosphere than they are at sea-level (Masarik and atoms, so 7Be and 10Be adsorb rapidly to organic and Beer 1999, 2009). Most of the production of cosmo- inorganic solid phases in the terrestrial and marine envi- genic Be therefore occurs in the upper atmosphere ronment. Thus, cosmogenic isotopes of beryllium can be (5–30 km), although there is trace, but measurable used to quantify surface age, sediment source, mixing production as oxygen atoms in minerals at the Earth’s rates, and particle residence and transit times in soils, surface are spallated (in situ produced; see Lal 2011, streams, lakes, and the oceans. -

12 Natural Isotopes of Elements Other Than H, C, O
12 NATURAL ISOTOPES OF ELEMENTS OTHER THAN H, C, O In this chapter we are dealing with the less common applications of natural isotopes. Our discussions will be restricted to their origin and isotopic abundances and the main characteristics. Only brief indications are given about possible applications. More details are presented in the other volumes of this series. A few isotopes are mentioned only briefly, as they are of little relevance to water studies. Based on their half-life, the isotopes concerned can be subdivided: 1) stable isotopes of some elements (He, Li, B, N, S, Cl), of which the abundance variations point to certain geochemical and hydrogeological processes, and which can be applied as tracers in the hydrological systems, 2) radioactive isotopes with half-lives exceeding the age of the universe (232Th, 235U, 238U), 3) radioactive isotopes with shorter half-lives, mainly daughter nuclides of the previous catagory of isotopes, 4) radioactive isotopes with shorter half-lives that are of cosmogenic origin, i.e. that are being produced in the atmosphere by interactions of cosmic radiation particles with atmospheric molecules (7Be, 10Be, 26Al, 32Si, 36Cl, 36Ar, 39Ar, 81Kr, 85Kr, 129I) (Lal and Peters, 1967). The isotopes can also be distinguished by their chemical characteristics: 1) the isotopes of noble gases (He, Ar, Kr) play an important role, because of their solubility in water and because of their chemically inert and thus conservative character. Table 12.1 gives the solubility values in water (data from Benson and Krause, 1976); the table also contains the atmospheric concentrations (Andrews, 1992: error in his Eq.4, where Ti/(T1) should read (Ti/T)1); 2) another category consists of the isotopes of elements that are only slightly soluble and have very low concentrations in water under moderate conditions (Be, Al). -

Production and Evolution of Li, Be and B Isotopes in the Galaxy
Astronomy & Astrophysics manuscript no. Prantzos˙Libeb2 c ESO 2018 July 18, 2018 Production and evolution of Li, Be and B isotopes in the Galaxy N.Prantzos1 Institut d’Astrophysique de Paris, UMR7095 CNRS, Univ.P. & M.Curie, 98bis Bd. Arago, 75104 Paris, France; e-mail: [email protected] ABSTRACT Context. We reassess the problem of the production and evolution of the light elements Li, Be and B and of their isotopes in the Milky Way in the light of new observational and theoretical developments. Aims. The main novelty is the introduction of a new scheme for the origin of Galactic cosmic rays (GCR), which for the first time enables a self-consistent calculation of their composition during galactic evolution. Methods. The scheme accounts for key features of the present-day GCR source composition, it is based on the wind yields of the Geneva models of rotating, mass-losing stars and it is fully coupled to a detailed galactic chemical evolution code. Results. We find that the adopted GCR source composition accounts naturally for the observations of primary Be and helps understanding why Be follows Fe more closely than O. We find that GCR produce ∼70% of the solar 11B/10B isotopic ratio; the remaining 30% of 11B presumably result from ν-nucleosynthesis in massive star explosions. We find that GCR and primordial nucleosynthesis can produce at most ∼30% of solar Li. At least half of the solar Li has to originate in low-mass stellar sources (red giants, asymptotic giant branch stars, or novae), but the required average yields of those sources are found to be much higher than obtained in current models of stellar nucleosynthesis. -
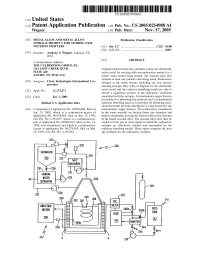
(12) Patent Application Publication (10) Pub. No.: US 2005/0254988A1 Wagner (43) Pub
US 2005O254988A1 (19) United States (12) Patent Application Publication (10) Pub. No.: US 2005/0254988A1 Wagner (43) Pub. Date: Nov. 17, 2005 (54) METALALLOY AND METALALLOY Publication Classification STORAGE PRODUCT FOR STORING EAST NEUTRON EMITTERS (51) Int. Cl. ................................................. C22C 43/00 (52) U.S. Cl. .................................................................. 420/1 (75) Inventor: Anthony S. Wagner, Lakeway, TX (US) (57) ABSTRACT Correspondence Address: THE CULBERTSON GROUP, PC. 1114 LOST CREEK BLVD. Aliquid reactant metal alloy includes at least one chemically SUTE 420 active metal for reacting with non-radioactive material in a AUSTIN, TX 78746 (US) mixed waste Stream being treated. The reactant alloy also includes at least one radiation absorbing metal. Radioactive (73) Assignee: Clean Technologies International Cor isotopes in the waste Stream, including any fast neutron poration emitting isotopes alloy with, or disperse in, the chemically Appl. No.: 11/173,271 active metal and the radiation absorbing metals are able to (21) absorb a significant portion of the radioactive emissions (22) Filed: Jul. 1, 2005 asSociated with the isotopes. A transmutation target fraction is included for absorbing fast neutrons and a transmutation Related U.S. Application Data emission absorbing fraction is provided for absorbing emis sions that result from the absorption of a fast neutron by the (63) Continuation of application No. 10/059,808, filed on transmutation target fraction. Non-radioactive constituents Jan. 29, 2002, which is a continuation-in-part of in the waste material are broken down into harmless and application No. 09/334,985, filed on Jun. 17, 1999, useful constituents, leaving the alloyed radioactive isotopes now Pat. -

Murchison Meteorite
Lifetimes of interstellar dust from cosmic ray exposure ages of presolar silicon carbide Philipp R. Hecka,b,c,1, Jennika Greera,b,c, Levke Kööpa,b,c, Reto Trappitschd, Frank Gyngarde,f, Henner Busemanng, Colin Madeng, Janaína N. Ávilah, Andrew M. Davisa,b,c,i, and Rainer Wielerg aRobert A. Pritzker Center for Meteoritics and Polar Studies, The Field Museum of Natural History, Chicago, IL 60605; bChicago Center for Cosmochemistry, The University of Chicago, Chicago, IL 60637; cDepartment of the Geophysical Sciences, The University of Chicago, Chicago, IL 60637; dNuclear and Chemical Sciences Division, Lawrence Livermore National Laboratory, Livermore, CA 94550; ePhysics Department, Washington University, St. Louis, MO 63130; fCenter for NanoImaging, Harvard Medical School, Cambridge, MA 02139; gInstitute of Geochemistry and Petrology, ETH Zürich, 8092 Zürich, Switzerland; hResearch School of Earth Sciences, The Australian National University, Canberra, ACT 2601, Australia; and iEnrico Fermi Institute, The University of Chicago, Chicago, IL 60637 Edited by Mark H. Thiemens, University of California San Diego, La Jolla, CA, and approved December 17, 2019 (received for review March 15, 2019) We determined interstellar cosmic ray exposure ages of 40 large ago. These grains are identified as presolar by their large isotopic presolar silicon carbide grains extracted from the Murchison CM2 anomalies that exclude an origin in the Solar System (13, 14). meteorite. Our ages, based on cosmogenic Ne-21, range from 3.9 ± Presolar stardust grains are the oldest known solid samples 1.6 Ma to ∼3 ± 2 Ga before the start of the Solar System ∼4.6 Ga available for study in the laboratory, represent the small fraction ago. -

IS* International Cosmic Ray Conference
IS* International Cosmic Ray Conference VOLUME a PLOVDIV* "*V ,W* BULGARIA AUGUST 13-86,1377 15" International Cosmic Ray Conference CONFERENCE PAPERS VOLUME a OG SESSION BULGARIAN ACADEMY OF SCIENCES PLOVDIV. BULGARIA AUGUST 13-26,19(77- Ill PREFACE The present publication contains the proceedings of the 15th International Cosmic Ray Confe- rence, Plovdiv, 13-26 August, 1977. This Conference is to be held under the auspices of the Inter- national Union of Pure and Applied Physics, organized by the Bulgarian Academy of Sciences. The publication comprises 12 volumes. Volumes from 1 to 9 include the original contribu- tions, which have arrived at the Secretariat of the National Organizing Committee by May 26, 1977. Papers which have been declared but not submitted by that date have been represented by their abstracts. Volumes from 10 to 12 include the invited and rapporteur lectures, as well as late origi- nal papers. Volume 12 contains the general contents of the volumes, an authors' index and other references. All papers included in the present publication are exact reproductions of the authors' original manuscripts. The Secretariat has not made any corrections or changes in the texts. The original contributions have been accepted and included in the programme after a decision of the Interna- tional Programme Advisory Board of the 15th ICRC on the basis of their abstracts. The full texts of the papers, however, have not been refereed by the editorial board of the present publication. The first nine volumes have been organized in accordance with the classical headings adopted at the cosmic ray conferences, which also coincide with the sessions. -

Nucleosynthesis •Light Element Processes •Neutron Capture Processes •LEPP and P Nuclei •Explosive C-Burning •Explosive H-Burning •Degenerate Environments
Lecture 21: Nucleosynthesis •Light element processes •Neutron capture processes •LEPP and P nuclei •Explosive C-burning •Explosive H-burning •Degenerate environments Lecture 21: Ohio University PHYS7501, Fall 2017, Z. Meisel ([email protected]) Nuclear Astrophysics: Nuclear physics from dripline to dripline •The diverse sets of conditions in astrophysical environments leads to a variety of nuclear reaction sequences •The goal of nuclear astrophysics is to identify, reduce, and/or remove the nuclear physics uncertainties to which models of astrophysical environments are most sensitive A.Arcones et al. Prog.Theor.Part.Phys. (2017) 2 In the beginning: Big Bang Nucleosynthesis • From the expansion of the early universe and the cosmic microwave background (CMB), we know the initial universe was cool enough to form nuclei but hot enough to have nuclear reactions from the first several seconds to the first several minutes • Starting with neutrons and protons, the resulting reaction S. Weinberg, The First Three Minutes (1977) sequence is the Big Bang Nucleosynthesis (BBN) reaction network • Reactions primarily involve neutrons, isotopes of H, He, Li, and Be, but some reaction flow extends up to C • For the most part (we’ll elaborate in a moment), the predicted abundances agree with observations of low-metallicity stars and primordial gas clouds • The agreement between BBN, the CMB, and the universe expansion rate is known as the Concordance Cosmology Coc & Vangioni, IJMPE (2017) 3 BBN open questions & Predictions • Notable discrepancies exist between BBN predictions (using constraints on the astrophysical conditions provided by the CMB) and observations of primordial abundances • The famous “lithium problem” is the several-sigma discrepancy in the 7Li abundance. -

NUCLEAR GEOMETRY: from HYDROGEN to BORON Alexander I
Materials Physics and Mechanics 45 (2020) 132-149 Received: March 11, 2020 NUCLEAR GEOMETRY: FROM HYDROGEN TO BORON Alexander I. Melker St. Petersburg Academy of Sciences on Strength Problems, Peter the Great St. Petersburg Polytechnic University Polytekhnicheskaya 29, 195251, St. Petersburg, Russian Federation e-mail: [email protected] Abstract. Possible ways of nuclear synthesis in the range from hydrogen to boron are studied. The geometric model of these nuclei is suggested. The basis for this model is the analogy 4 4 between tetrahedral fullerene C4 and helium 2He . It is assumed that a nucleus of helium 2He has the form of a tetrahedron, where: 1) All the apices are equivalent and therefore they are protons, 2) Each neutron in a nucleus decomposes into a proton and three negatively charged particles having the charge ⅓ of that of an electron, 3) Interaction of the negative particles creates a special electronic pattern, which symmetry does not coincide with that of protons one, but determines it. On the basis of the postulates, the structure of other nuclei has been designed using geometric modeling. For hydrogen, deuterium, tritium and helium 3, a point, a linear and a plane structure respectively have been obtained. Helium 4 has tetrahedral symmetry. Then there was transition from three-fold symmetry prisms (lithium 6 and 7) to five-fold symmetry (boron 10 and 11) through four-fold one (beryllium 8, 9, 10). The nuclear electron patterns are more complex; their polyhedrons resemble the electron pairs arrangement at the valence shells of molecules. Keywords: beryllium, boron, deuterium, graph representation, helium, hydrogen, lithium, nuclear electron, nuclear geometry 1. -

P4 ATOMIC STRUCTURE Question Practice Class: ______Date: ______
Name: ________________________ P4 ATOMIC STRUCTURE Question Practice Class: ________________________ Date: ________________________ Time: 132 minutes Marks: 129 marks Comments: HIGHER TIER Page 1 of 44 (a) The graph shows how the count rate from a sample containing the radioactive substance 1 cobalt-60 changes with time. (i) What is the range of the count rate shown on the graph? From __________ counts per second to __________ counts per second. (1) (ii) How many years does it take for the count rate to fall from 200 counts per second to 100 counts per second? Time = _________________________ years (1) (iii) What is the half-life of cobalt-60? Half-life = _________________________ years (1) Page 2 of 44 (b) The gamma radiation emitted from a source of cobalt-60 can be used to kill the bacteria on fresh, cooked and frozen foods. Killing the bacteria reduces the risk of food poisoning. The diagram shows how a conveyor belt can be used to move food past a cobalt-60 source. (i) Which one of the following gives a way of increasing the amount of gamma radiation the food receives? Put a tick ( ) in the box next to your answer. Increase the temperature of the cobalt-60 source. Make the conveyor belt move more slowly. Move the cobalt-60 source away from the conveyor belt. (1) (ii) To protect people from the harmful effects of the gamma radiation, the cobalt-60 source has thick metal shielding. Which one of the following metals should be used? Draw a ring around your answer. aluminium copper lead (1) Page 3 of 44 (c) A scientist has compared the vitamin content of food exposed to gamma radiation with food that has not been exposed. -

Static Properties of Neutron-Rich and Proton-Rich Isotopes
Physical Science & Biophysics Journal MEDWIN PUBLISHERS ISSN: 2641-9165 Committed to Create Value for Researchers Static Properties of Neutron-Rich and Proton-Rich Isotopes Zari Binesh* Research Article Department of Nuclear Physics, Faculty of Physics and Nuclear Engineering, Shahrood Volume 4 Issue 2 University of Technology, Shahrood, Iran Received Date: September 28, 2020 Published Date: October 28, 2020 *Corresponding author: Zari Binesh, Department of Nuclear Physics, Faculty of Physics and Nuclear Engineering, Shahrood University of Technology, Shahrood, Iran, Tel: 09102878009; Email: [email protected] Abstract In this paper, we have investigated some static properties of 9Be, 9B, 13C, 13N, 17O, 17F, 21Ne, and 21Na isotopes. The ground state and excited state energy of these isotopes have determined in the relativistic shell model and compared with experimental data. The calculated charge radius of them is in good agreement with experimental results. Keywords: Charge radius; Energy levels Introduction There are two stable isotopes of carbon: 12C and 13C. 13C is used for instance in organic chemistry research, studies Investigating neutron-rich or proton-rich isotopes are into molecular structures, metabolism, food labeling, air one of the interesting subjects in nuclear science. These pollution and climate change. It is also used in breath tests isotopes have some single nucleon out of the closed core in to determine the presence of the helicobacter pylori bacteria shell structure [1]. Determining the energy levels, charge which causes stomach ulcer. 13C can also be used for the radius and other static properties of nuclei, is one of the production of the radioisotope 13N which is a PET isotope useful components to cognition the nuclear structure [2].