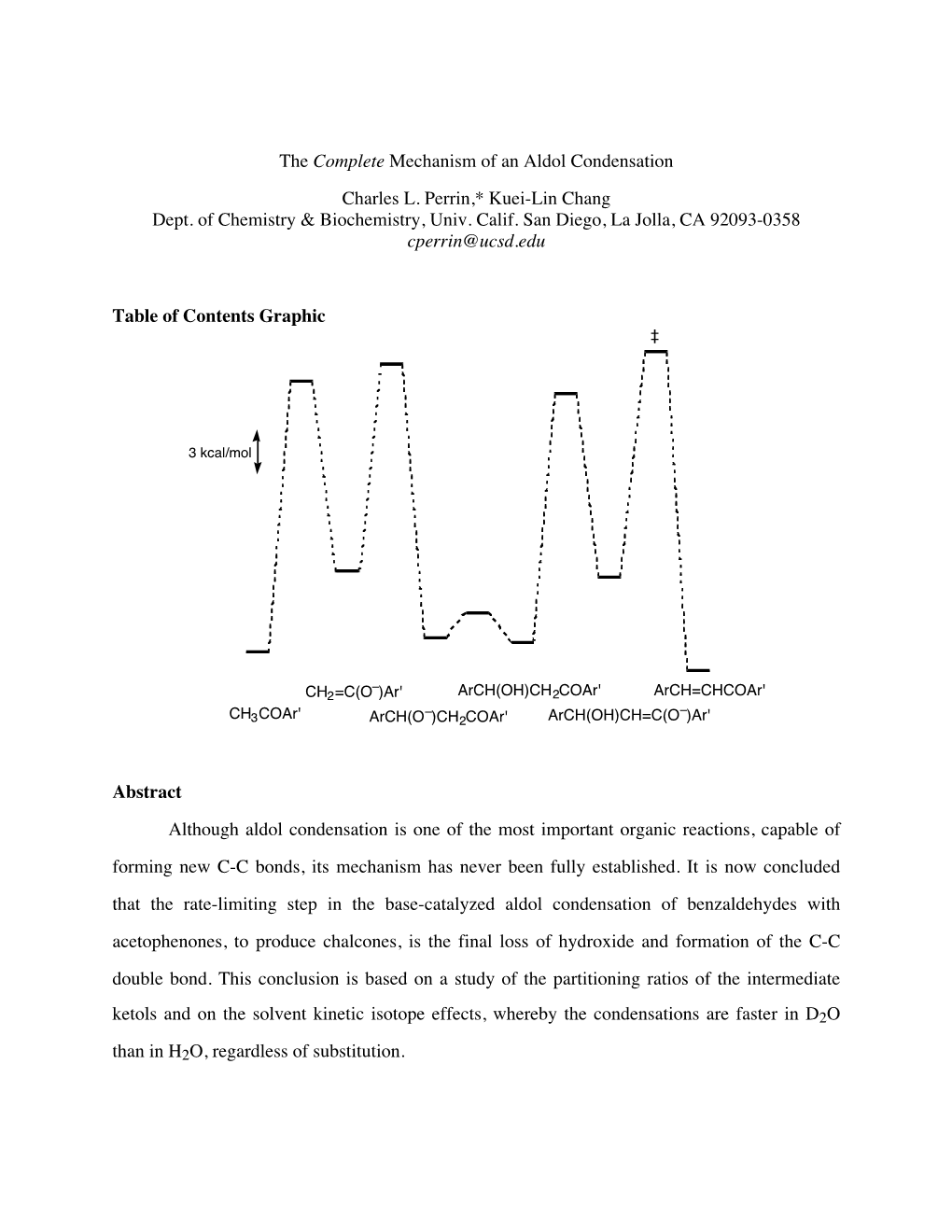
The Complete Mechanism of an Aldol Condensation Charles L. Perrin,* Kuei-Lin Chang Dept
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Diversity-Oriented Combinatorial Biosynthesis of Benzenediol Lactone Scaffolds by Subunit Shuffling of Fungal Polyketide Synthases
Diversity-oriented combinatorial biosynthesis of benzenediol lactone scaffolds by subunit shuffling of fungal polyketide synthases Yuquan Xua,b,1, Tong Zhouc,1, Shuwei Zhangc, Patricia Espinosa-Artilesb, Luoyi Wangc, Wei Zhanga, Min Lina, A. A. Leslie Gunatilakab,d, Jixun Zhanc,2, and István Molnárb,d,2 aBiotechnology Research Institute, The Chinese Academy of Agricultural Sciences, Beijing 100081, P. R. China; bNatural Products Center, School of Natural Resources and the Environment, University of Arizona, Tucson, AZ 85706; cDepartment of Biological Engineering, Utah State University, Logan, UT 84322; and dBio5 Institute, University of Arizona, Tucson, AZ 85721 Edited by Jerrold Meinwald, Cornell University, Ithaca, NY, and approved June 23, 2014 (received for review April 16, 2014) Combinatorial biosynthesis aspires to exploit the promiscuity of biosynthesis inhibitory activities in animals. 10,11-dehydrocurvularin microbial anabolic pathways to engineer the synthesis of new (7;Fig.1)isaDALwitha12-memberedring(DAL12)that chemical entities. Fungal benzenediol lactone (BDL) polyketides modulates the heat shock response and the immune system (8, 9). are important pharmacophores with wide-ranging bioactivities, BDL scaffolds are biosynthesized by pairs of collaborating, including heat shock response and immune system modulatory sequentially acting iterative polyketide synthases (iPKSs) (3) – effects. Their biosynthesis on a pair of sequentially acting iterative forming quasi-modular BDL synthases (BDLSs) (Fig. 1) (11 14). polyketide synthases (iPKSs) offers a test case for the modulariza- Each of the BDLS subunits catalyze recursive, decarboxylative tion of secondary metabolic pathways into “build–couple–pair” Claisen condensations of malonyl-CoA using a single core set of ketoacyl synthase (KS), acyl transferase (AT), and acyl carrier combinatorial synthetic schemes. -

Experiment 19 — Aldol Condensation
Chem 22 Spring 2010 Experiment 19 — Aldol Condensation _____________________________________________________________________________ Pre-lab preparation. (1) Write the mechanism of the base-catalyzed aldol condensation of acetone and a generalized aromatic aldehyde, Ar–CH=O to give the α,β-unsaturated product (i.e. the reaction at the top of the next page, but with a 1:1 ratio of ketone and aldehyde). Remember that dehydration in this case occurs under basic conditions, so it can't start with protonation of the hydroxyl group. Nor can it go via an E2 pathway. (2) Draw the structures of all the possible aldehyde and ketone reactants (not the 25 possible condensation products!). (3) The new CC double bonds of the condensation products are E rather than Z, as shown in the acetone example. Why? (4) What's the purpose of rinsing the crude product with dilute acetic acid, followed by ethanol? (5) What is the procedure for carrying out a single-solvent recrystallization? Write this out in detail. The aldol condensation has historically been one of the favorite tools in the synthetic organic chemist's repertoire because of its versatility in forming new CC bonds. Since its discovery in the 1870s the aldol condensation has been use extensively in the synthesis of natural products and other complex molecules. In a typical base-catalyzed aldol condensation an enolate ion attacks the carbonyl group of an aldehyde or ketone. This carbonyl addition produces a β-hydroxy carbonyl compound. In many cases the initially formed condensation product undergoes an "E1cB" dehydration to produce an α,β-unsaturated carbonyl compound as the final product. -

Aldol Condensation- Aldehyde (Or Ketone) Enolate Condenses with Aldehyde (Or Ketone)
Chem 232 Summary of Alpha Substitutions page 1 Aldol Condensation- aldehyde (or ketone) enolate condenses with aldehyde (or ketone): O CH O O H 3 H CH 3 CH3 C C C C CH C C CH2 CH2 H -H O H H O OH 2 H nucleophile electrophile -hydroxy aldehyde -unsaturated aldehyde The nucleophile can be a ketone enolate or aldehyde enolate and the electrophile (shaded) can be an aldehyde or ketone. Crossed Aldol- Condensation between two different carbonyls. The component without hydrogens is the electrophile: O O O OH O C CH C C CH CH3 C H CH -H2O CH 2 CH3 H 3 ketone enolate no -hydrogens -unsaturated ketone Aldol Cyclizations- A dicarbonyl produces an enolate and carbonyl in the same molecule: enolate from a O OH 1,5-diketone CH OH 3 CH3 O O -H2O O CH2 CH3 CH3 CH2 O Claisen Condensation- Similar to Aldol condensation except the nucleophile is an ester enolate; O O O O O O + EtO C CH C OEt EtO C CH2 C EtO C CH2 CH3 C OEt 2 ketoester CH3 CH3 Dieckmann Cyclization- Internal Claisen condensation similar to Aldol cyclization. A 1,6 diester gives a 5-membered ring and a 1,7 diester gives a 6-membered ring: O OEt O OEt cyclic ketoester C C OEt O O Crossed Claisen- Similar to crossed Aldol- Electrophile has nohydrogens: O O O O C C EtO EtO C CH2 EtO C CH2 ketoester Variations on Crossed Claisen- ketone enolate and ester condensation. Esters, carbonates, formates and oxalates are common electrophiles: O O O O O O O O H C OEt H EtO C OEt OEt ethyl formate -ketoaldehyde diethyl carbonate -ketoester O O O O O OEt diketoester EtO C C OEt O diethyl oxalate -

Aldol Condensation
Chemistry 212 Laboratory Dibenzalacetone via Crossed Aldol Condensation Prelab: Calculate the amounts of all chemicals needed in measurable amounts (i.e. grams or milliliters rather than moles.) Introduction: Aldol condensations are important in organic synthesis, providing a good way to form carbon–carbon bonds. The "aldol" (aldehyde + alcohol) product is a structural unit found in many naturally occurring molecules and pharmaceuticals, and is therefore important. In an Aldol condensation an enolate ion reacts with a carbonyl compound to form a β- hydroxyaldehyde or β-hydroxyketone, followed by dehydration to give a conjugated enone. The general equation is shown in Figure 1. O O O R" B: H R R'" R "R R'" loss of H2O H R' R' Figure 1. The equation for the Aldol Condensation. The reaction involves the nucleophilic addition of an enolate to an aldehyde to form a β-hydroxy carbonyl. The β-hydroxy carbonyl is readily dehydrated under mild conditions. The aldol reaction occurs under both acidic and basic conditions as seen in Figure 2. ENOL pathway (reacts in H O protonated OH form) O O catalytic H+ O O H H R' H2O lost R' R R R' R R H aldol addition product aldol condensation product ENOLATE pathway O O M O M O base O H R' R R' R R enolate H Figure 2. The Aldol reaction and subsequent dehydration under acidic and basic conditions. The reaction we will be doing this week involves the reaction between benzaldehyde and acetone to do a double Aldol Condensation. The overall equation is shown in Figure 3. -

Biocatalyzed Synthesis of Statins: a Sustainable Strategy for the Preparation of Valuable Drugs
catalysts Review Biocatalyzed Synthesis of Statins: A Sustainable Strategy for the Preparation of Valuable Drugs Pilar Hoyos 1, Vittorio Pace 2 and Andrés R. Alcántara 1,* 1 Department of Chemistry in Pharmaceutical Sciences, Faculty of Pharmacy, Complutense University of Madrid, Campus de Moncloa, E-28040 Madrid, Spain; [email protected] 2 Department of Pharmaceutical Chemistry, Faculty of Life Sciences, Althanstrasse 14, A-1090 Vienna, Austria; [email protected] * Correspondence: [email protected]; Tel.: +34-91-394-1823 Received: 25 February 2019; Accepted: 9 March 2019; Published: 14 March 2019 Abstract: Statins, inhibitors of 3-hydroxy-3-methylglutaryl coenzyme A (HMG-CoA) reductase, are the largest selling class of drugs prescribed for the pharmacological treatment of hypercholesterolemia and dyslipidaemia. Statins also possess other therapeutic effects, called pleiotropic, because the blockade of the conversion of HMG-CoA to (R)-mevalonate produces a concomitant inhibition of the biosynthesis of numerous isoprenoid metabolites (e.g., geranylgeranyl pyrophosphate (GGPP) or farnesyl pyrophosphate (FPP)). Thus, the prenylation of several cell signalling proteins (small GTPase family members: Ras, Rac, and Rho) is hampered, so that these molecular switches, controlling multiple pathways and cell functions (maintenance of cell shape, motility, factor secretion, differentiation, and proliferation) are regulated, leading to beneficial effects in cardiovascular health, regulation of the immune system, anti-inflammatory and immunosuppressive properties, prevention and treatment of sepsis, treatment of autoimmune diseases, osteoporosis, kidney and neurological disorders, or even in cancer therapy. Thus, there is a growing interest in developing more sustainable protocols for preparation of statins, and the introduction of biocatalyzed steps into the synthetic pathways is highly advantageous—synthetic routes are conducted under mild reaction conditions, at ambient temperature, and can use water as a reaction medium in many cases. -

Cross-Aldol Condensation of Acetone and N-Butanol Into Aliphatic Ketones Over Supported Cu Catalysts on Ceria-Zirconia
catalysts Article Cross-Aldol Condensation of Acetone and n-Butanol into Aliphatic Ketones over Supported Cu Catalysts on Ceria-Zirconia Minseok Kim 1, Jongha Park 1, Hari Prasad Reddy Kannapu 1,2 and Young-Woong Suh 1,2,* ID 1 Department of Chemical Engineering, Hanyang University, Seoul 04763, Korea; [email protected] (M.K.); [email protected] (J.P.); [email protected] (H.P.R.K.) 2 Research Institute of Industrial Science, Hanyang University, Seoul 04763, Korea * Correspondence: [email protected]; Tel.: +82-2-2220-2329 Academic Editor: Christophe Len Received: 7 August 2017; Accepted: 23 August 2017; Published: 24 August 2017 Abstract: A long-chain hydrocarbon biofuel of jet fuel range can be produced via aldol condensation of fermented products such as acetone and alcohols over the catalysts containing both metallic sites for the dehydrogenation of alcohols and basic sites for the condensation reaction. However, an efficient catalyst system has not been studied widely yet the route is promising for biofuel production. In this work, Cu catalysts supported on ceria-zirconia (Cu/xCeZr) were prepared using coprecipitated CexZr1-xO2 supports with different Ce/Zr ratios for the cross-aldol condensation of acetone and n-butanol into mono- and di-alkylated aliphatic ketones, 2-heptanone and 6-undecanone. The acetone conversion and 6-undecanone selectivity increased with specific Cu surface area due to formation of the dehydrogenation product butyraldehyde at a higher concentration. The total yield of cross-aldol condensation products was strongly dependent on a combination of Cu sites and basic sites. This was confirmed by the results in the reaction between acetone and butyraldehyde over supported Cu catalysts that additionally examined the adsorbed acyl species on Cu surface taking part in the aldol condensation reaction. -

THE VIRTUAL MIXED ALDOL REACTION in This Virtual Experiment, You Will Use the Virtual Laboratory to Synthesize a Beta-Hydroxy Ketone Using a Mixed Aldol Reaction
THE VIRTUAL MIXED ALDOL REACTION In this virtual experiment, you will use the virtual laboratory to synthesize a beta-hydroxy ketone using a mixed aldol reaction. By making modifications to the reaction conditions, you will try to find conditions that will allow for the elimination of water in an aldol condensation reaction. Introduction The aldol reaction is an important C-C bond forming reaction that joins two carbonyl containing compounds together by using a base. The first step in the aldol reaction is the deprotonation of the α-hydrogen (note: α refers to the carbon position next to a C=O bond, β refers to the position 2 carbons away from the carbonyl). The hydrogens at the α position are slightly acidic due to the resonance stabilization of the corresponding enolate (aldehyde pKa = 17; ketone pKa= 20). The high pKa values indicate that in order to deprotonate the α-hydrogen of an aldehyde, an extremely strong base is required. This deprotonation was initially done using bases such as potassium hydroxide, (pKa of water, the conjugate acid of hydroxide = 15.7) which would only deprotonate 5% of the aldehydes (1015.7/1017) and 0.005% of the ketones (1015.7/1020). The aldol reaction was made a little more efficient by using a slightly stronger base such as potassium t-butoxide (pKa of t-butanol, its conjugate acid = 17). Like potassium hydroxide, potassium t-butoxide ionizes a small percentage of the carbonyl compound to the enolate ion. Complete and irreversible deprotonation requires a superbase such as lithium diisopropylamide (LDA, pKa of it’s conjugate acid = 35.7). -

The Aldol Condensation
R Carbon The Aldol Condensation R Carbon Carbon Contents Objectives 1 Introduction 1 Safety 1 Preparation of 4-(4’-methoxyphenyl)-3-buten-2-one (Product A) 2 Preparation of 1,5-bis(4’-methoxyphenyl)-1,4-pentadien-3-one (Product B) 6 Manuscript prepared by Dr. A. Jonathan Singh and Dr. Hemi Cumming. School of Chemical and Physical Sciences, Victoria University of Wellington, New Zealand. R Carbon Objectives In this experiment, the aldol condensation of acetone and p-anisaldehyde The objective of this experiment is to understand (4-methoxybenzaldehyde) is carried out under basic aspects of carbonyl chemistry and carbon-carbon conditions (Scheme 2). By employing a stepwise bond formations using the well-known aldol sequence, you will be able to isolate the mono- condensation reaction. Reaction products formed addition (Product A), and repeat the reaction, this in this experiment will be primarily characterized time using (Product A) as the source ketone to 1 by H NMR spectroscopy using the Spinsolve form the bis-addition (Product B). benchtop NMR spectrometer. Safety Introduction This experiment must be performed in a fume hood with adequate ventilation. Acetophenone Carbon-carbon bond formation is one of the and benzaldehyde are harmful – handle with care. cornerstones of organic synthesis. One of the Potassium hydroxide is caustic and corrosive – use key reactions used, the aldol condensation, with caution. Wear appropriate safety equipment features the reaction of two carbonyl compounds before commencing with this experiment. to form a new β-hydroxy carbonyl compound.1 Consult the relevant MSDS for additional safety This reaction can be performed under acid- or information. -

Chem 353: Aldol Condensation
ALDOL.1 ORGANIC SYNTHESIS: ALDOL CONDENSATION REACTION TECHNIQUES REQUIRED: Filtration (Vacuum), Recrystallisation, Melting Point Determination, Yield calculation OTHER DOCUMENTS: Experimental Procedure, Report template (pdf), Report template (doc), Spectra INTRODUCTION In an "aldol addition" reaction, an enol or more commonly an enolate of an aldehyde or ketone reacts with a second aldehyde or ketone forming a new carbon-carbon bond. This makes the aldol reaction an important reaction for organic synthesis. Originally, the aldol reaction used ethanal (see below) and therefore the product contained both an aldehyde and an alcohol functional group; thus it became known as the aldol reaction. Recall that aldehydes and ketones typically undergo nucleophilic addition reactions. The aldol reaction demonstrates how carbonyl compounds can react as electrophiles or nucleophiles depending on the reaction conditions and what other species are present. Alcohols are similar in that respect. Can you think of examples to illustrate that ? The aldol reaction requires an aldehyde or ketone that contains at least one -hydrogen (the -hydrogen is on the carbon adjacent to the C=O group) since the -hydrogen is required in order to form the enol or enolate. In the base-catalysed aldol reaction, the relatively acidic hydrogen on the -carbon (typical pKa 16-20) is deprotonated by a base to form the enolate. The enolate reacts as a carbon nucleophile that can then react with the electrophilic carbonyl carbon of another aldehyde or ketone molecule. Depending on the strength of the base used, the extent of deprotonation can be controlled. If a strong base is used (such as lithium diisopropylamide, LDA) then deprotonation is quantitative (100%). -

Aldol Condensations of Aldehydes and Ketones Catalyzed by Primary Amine on Water
Asian Journal of Chemistry; Vol. 24, No. 2 (2012), 751-755 Aldol Condensations of Aldehydes and Ketones Catalyzed by Primary Amine on Water 1 1,* 1 1 2 1,* XU ZHANG , YAN XIONG , SHUTING ZHANG , XUEGE LING , JINYUE WANG and CHANGGUO CHEN 1College of Chemistry and Chemical Engineering, Chongqing University, Chongqing 400044, P.R. China 2Yibin Research Center of Chemical & Textile Industry, Key Laboratory of Computational Physics in Universities of Sichuan, Yibin University, Yibin 644000, P.R. China *Corresponding authors: E-mail: [email protected]; [email protected] (Received: 15 March 2011; Accepted: 14 October 2011) AJC-10521 Potassium glycinate-catalyzed aldol condensation reactions of aromatic aldehydes and ketones on water at room temperature have been developed. Under optimal conditions, various condensation adducts are furnished in up to 63 % yield. By simple separation of the oil phase, potassium glycinate-containing water is reused to catalyze aldol condensation for six runs without loss of catalytic activity. Theoretical investigation reveals correlation between the yields and dehydroxylation energy barriers of aldol products, reasonably low activation energy of 11.9 kcal/mol for transition state formation in condensation of benzaldehyde and acetone, vibrations between aldol donor and acceptor, which correspond to the only imaginary frequency (-179.8i). Key Words: Aldol condensations, Glycine, Heterogeneous catalysis, Primary amine, ααα,βββ-Unsaturated ketones. INTRODUCTION sation reactions with equimolar water as cocatalyst13. Proline- derived organocatalysts are also presented to facilitate aldol The aldol condensation reaction has been applied to C-C condensations, in which secondary amine readily forming bond formation in synthetic organic chemistry and is recognized corresponding active enamines are underlined14-17. -

Solvent-Free Organic Synthesis
Chem. Rev. 2000, 100, 1025−1074 1025 Solvent-Free Organic Synthesis K. Tanaka† and F. Toda*,‡ Department of Applied Chemistry, Faculty of Engineering, Ehime University, Matsuyama, Ehime 790-8577, Japan, and Department of Chemistry, Faculty of Science, Okayama University of Science, 1-1 Ridaicho, Okayama 700-0005, Japan Received June 17, 1996 Contents G. Enantioselective Photoreaction 1056 1. Enantioselective Photoreactions of Chiral 1056 I. Introduction 1025 Molecules II. Molecular Movement in the Solid State 1026 2. Enantioselective Photoreaction of Achiral 1057 III. Thermal Reaction 1028 Molecules in Chiral Inclusion Crystals A. Oxidation 1028 3. Enantioselective Photoreaction of Achiral 1066 B. Reduction 1030 Molecules in Their Chiral Crystals C. Addition Reaction 1032 V. Conclusion 1071 1. Halogenation and Hydrohalogenation 1032 VI. References 1071 2. Michael Addition and Aldol Addition 1032 D. Elimination Reaction 1034 I. Introduction E. C−C Coupling Reaction 1034 Crushed grapes give wine by fermentation, but 1. [2+2], [4+2], and [6+2] Cycloaddition 1034 dried grapes do not result in wine. Although milk Reaction turns sour and shaking of milk gives cheese, dried 2. Aldol Condensation Reaction 1036 milk can be kept unaltered. Similarly dried meat can 3. Dieckmann Condensation Reaction 1037 be stored for a long time, whereas meat soup rapidly 4. Grignard, Reformatsky, and Luche 1037 putrefies on standing. Reactions By observation of these phenomena, one can see 5. Wittig Reaction 1038 that conversion of one material into another one 6. Ylid Reaction 1038 occurs in the liquid state but not in the solid state. 7. Pinacol Coupling Reaction 1039 One of the most famous ancient philosophers in 8. -

Organic Synthesis Under Solvent-Free Condition: an Environmentally Benign Procedure- I
View metadata, citation and similar papers at core.ac.uk brought to you by CORE provided by ePrints@Bangalore University GENERAL [ ARTICLE Organic Synthesis under Solvent-free Condition: An Environmentally Benign Procedure- I Gopalpur Nagendrappa Though it is a common practice to run the organic reactions in solvent media, the chemists' concern to minimize the environmental pollution caused by solvents and also their academic interest in solid-solid reactions have led them in recent times to develop methodologies for solvent-free reac- The author is a Professor tions with considerable success. of Organic Chemistry at Bangalore University, The Function of a Solvent Bangalore. His main area of research is A general assumption with regard to organic reactions is that organosilicon chemistry with particular attention they are performed in a solvent medium. The rationale behind to developing new this concept is simple. That is, the reactants can interact effec- synthetic procedures and tively if they are in a homogeneous solution, which facilitates reagents, and studying the the stirring, shaking or other ways of agitation, whereby the reaction mechanisms. reactant molecules come together rapidly and continuously. Moreover, uniform heating or cooling of the mixture, if needed, can be carried out in a solution relatively easily. However, the role of a solvent in the context of an organic reaction is much more complex than merely providing a homogeneous setting for a large number of collisions of the reactants to take place. A solvent has the power to enhance or reduce the speed of a reaction, at times enormously. Changing of solvent of a reaction can influence the rate of that reaction, and it can be powerful enough to change the reaction course itself.