De Novo Transcriptome Assembly Databases for the Butterfly Orchid
Total Page:16
File Type:pdf, Size:1020Kb

Load more
Recommended publications
-

An Integrated Orchid Functional Genomics Database
Orchidstra: An Integrated Orchid Functional Genomics Database Special Focus Issue Chun-lin Su1,3, Ya-Ting Chao1,3, Shao-Hua Yen1, Chun-Yi Chen1, Wan-Chieh Chen1, Yao-Chien Alex Chang2 and Ming-Che Shih1,* 1Agricultural Biotechnology Research Center, Academia Sinica, Taipei 11529, Taiwan 2Department of Horticulture and Landscape Architecture, National Taiwan University, Taipei 10617, Taiwan. 3These authors contributed equally to this work. *Corresponding author: E-mail: [email protected]; Fax, +886-2-26515693. (Received November 9, 2012; Accepted January 5, 2013) A specialized orchid database, named Orchidstra (URL: Abbreviations: BLAST, basic local alignment search tool; – Databases http://orchidstra.abrc.sinica.edu.tw), has been constructed CAM, crassulacean acid metabolism; EIF5A, eukaryotic trans- to collect, annotate and share genomic information for lation initiation factor 5A; EST, expressed sequence tag; GO, orchid functional genomics studies. The Orchidaceae is a Gene Ontology; KEGG, Kyoto Encyclopedia of Genes and large family of Angiosperms that exhibits extraordinary bio- Genomes; miRNA, microRNA; NGS, next-generation sequen- diversity in terms of both the number of species and their cing; SRA, sequence read archive; TSA, transcriptome distribution worldwide. Orchids exhibit many unique biolo- shotgun assembly. gical features; however, investigation of these traits is cur- rently constrained due to the limited availability of genomic information. Transcriptome information for five orchid spe- Introduction cies and one commercial hybrid has been included in the Orchidaceae, the orchid family, diverged from the Liliaceae Orchidstra database. Altogether, these comprise >380,000 and Amaryllidaceae, is the largest family of Angiosperms, with non-redundant orchid transcript sequences, of which >800 genera and >25,000 species. -

Evolutionary Conservation of the Orchid MYB Transcription Factors DIV, RAD, and DRIF
ORIGINAL RESEARCH published: 01 November 2019 doi: 10.3389/fpls.2019.01359 Evolutionary Conservation of the Orchid MYB Transcription Factors DIV, RAD, and DRIF Maria Carmen Valoroso 1, Rómulo Sobral 2, Giuseppe Saccone 1, Marco Salvemini 1, Maria Manuela Ribeiro Costa 2 and Serena Aceto 1* 1 Department of Biology, University of Naples Federico II, Naples, Italy, 2 BioSystems & Integrative Sciences Institute (BioISI), Plant Functional Biology Centre, University of Minho, Campus de Gualtar, Braga, Portugal The MYB transcription factors DIVARICATA (DIV), DIV-and-RAD-Interacting-Factor (DRIF), and the small interfering peptide RADIALIS (RAD) can interact, forming a regulatory module that controls different plant developmental processes. In the snapdragon Antirrhinum majus, this module, together with the TCP transcription factor CYCLOIDEA (CYC), is responsible for the establishment of floral dorsoventral asymmetry. The spatial gene expression pattern of the OitDIV, OitDRIF, and OitRAD homologs of Orchis italica, an orchid with zygomorphic flowers, has suggested a possible conserved role of these Edited by: Jen-Tsung Chen, genes in bilateral symmetry of the orchid flower. Here, we have identified four DRIF genes National University of Kaohsiung, of orchids and have reconstructed their genomic organization and evolution. In addition, Taiwan we found snapdragon transcriptional cis-regulatory elements of DIV and RAD loci generally Reviewed by: Minsung Kim, conserved within the corresponding orchid orthologues. We have tested the biochemical University of Manchester, interactions among OitDIV, OitDRIF1, and OitRAD of O. italica, showing that OitDRIF1 United Kingdom Chao Bian, can interact both with OitDIV and OitRAD, whereas OitDIV and OitRAD do not directly Beijing Genomics Institute (BGI), interact, as in A. -

Molecularphylogeneticsof Phalaenopsis(Orchidaceae)
The JapaneseSocietyJapanese Society for Plant Systematics ISSN 1346-7565 Acta Phytotax. GeoboL 56 (2): 14]-161 (200S)・ Molecular Phylogenetics of Phalaenopsis (Orchidaceae)and allied Genera: Re-evaluation of Generic Concepts TOMOHISA YUKAWAi, KOICHI KITA2, TAKASHI HANDA2, TOPIK HIDAYAT3 and MOTOMHTo3 i71sukuba 21hstitute Botanical Garcien, Nlational Scienee Mtiseum, Amakuho, Tyuketba, 305-OO05. Jopan; of 3Graduate Agricultnre andforestn)). Uhivensity qf'Tgukuba, fennodai, 71yukuba, 305-857Z Japan; Schoot ofArts and Seience, Uhivensity of7bdy,o, Kbmaba, 7bkyo, J53-8902, JZu)an, Molecular phylogenetic analyscs were performed using data sets derived from DNA sequences ofthe plastid genome (matK and trnK introns) and the nuelear genome (rDNA ITS) in an examination ofrela- tionships of all sections ofPhataenqpsis and closely related gcnera. The fo11owing insights were pro- vided: (1) The genera Lesliea and IVbthodoritis are nested within Phalaenopsis, (2) Phalaenopsis subgenus Aphyilae and section EsmeJ'aldd, often treated as thc independent genera Kirrgidium and Doritis respectively, are also nested within Phalaenqpsis. (3) Two subgenera of Phalaenqpsis, namely, Phalaenopsis and 1larishianae, are not monophyletic. (4) Phalaenopsis sections Deliciosae, SZautqglottis, Amboinenses and Zehrinae are not monophyletic. (5) lnconsistencies bctween the plastid and nuclear lineages indicate a hybrid origin ofPhalaenopsis minus and Phalaenopsis phitmpinensis. (6) In light of these findings, and to accommodate phylogenetic integrity and stability in nomenclature, we adopt a broadly defincd Doritis characterized by the possession of fbur pollinia, an explicit character state. Key words: Doritis,introgression, ITS, mati(l moleculag Orchidaceae, Ahalaenopsis, phylogcnctics, tttnK Phakzenopsis Blume is an orchid genus to which 62 tion ofthe genus has been thoroughly reviewed by species are currently assigned (Christenson 2001). -

Nuclear DNA Contents of Phalaenopsis Sp. and Doritis Pulcherrima
J. AMER. SOC. HORT. SCI. 126(2):195–199. 2001. Nuclear DNA Contents of Phalaenopsis sp. and Doritis pulcherrima Sandy Lin and Hsiao-Ching Lee Department of Life Science, National Tsing Hua University, Hsinchu, 30043, Taiwan, Republic of China Wen-Huei Chen Department of Horticulture, Taiwan Sugar Research Institute, Tainan, 701, Taiwan, Republic of China Chi-Chang Chen and Yen-Yu Kao Department of Botany, National Taiwan University, Taipei, 10764, Taiwan, Republic of China Yan-Ming Fu and Yao-Huang Chen Department of Horticulture, Taiwan Sugar Research Institute, Tainan, 701, Taiwan, Republic of China Tsai-Yun Lin1 Department of Life Science, National Tsing Hua University, Hsinchu, 30043, Taiwan, Republic of China ADDITIONAL INDEX WORDS. Orchidaceae, endoreduplication, flow cytometry, genome size ABSTRACT. Nuclear DNA contents were estimated by flow cytometry in 18 Phalaenopsis Blume species and Doritis pulcherrima Lindl. DNA amounts differed 6.07-fold, from 2.74 pg/diploid nuclear DNA content (2C) in P. sanderiana Rchb.f. to 16.61 pg/2C in P. parishii Rchb.f. Nuclear DNA contents of P. aphrodite Rchb.f. clones, W01-38 (2n = 2x = 38), W01-41 (2n = 3x = 57), and W01-22 (2n = 4x = 76), displayed a linear relationship with their chromosome numbers, indicating the accuracy of flow cytometry. Our results also suggest that the 2C-values of the Phalaenopsis sp. correlate with their chromosome sizes. The comparative analyses of DNA contents may provide information to molecular geneticists and systematists for genome analysis in Phalaenopsis. Endoreduplication was found in various tissues of P. equestris at different levels. The highest degree of endoreduplication in P. -

Genome-Wide Identification and Classification of MIKC-Type MADS
He et al. BMC Plant Biology (2019) 19:223 https://doi.org/10.1186/s12870-019-1836-5 RESEARCH ARTICLE Open Access Genome-wide identification and classification of MIKC-type MADS-box genes in Streptophyte lineages and expression analyses to reveal their role in seed germination of orchid Chunmei He1, Can Si1,2, Jaime A. Teixeira da Silva3, Mingzhi Li4 and Jun Duan1* Abstract Background: MADS-box genes play crucial roles in plant floral organ formation and plant reproductive development. However, there is still no information on genome-wide identification and classification of MADS-box genes in some representative plant species. A comprehensive investigation of MIKC-type genes in the orchid Dendrobium officinale is still lacking. Results: Here we conducted a genome-wide analysis of MADS-box proteins from 29 species. In total, 1689 MADS-box proteins were identified. Two types of MADS-boxgenes,termedtypeIandII,werefoundinland plants, but not in liverwort. The SQUA, DEF/GLO, AG and SEP subfamilies existed in all the tested flowering plants, while SQUA was absent in the gymnosperm Ginkgo biloba, and no genes of the four subfamilies were found in a charophyte, liverwort, mosses, or lycophyte. This strongly corroborates the notion that clades of floral organ identity genes led to the evolution of flower development in flowering plants. Nine subfamilies of MIKCC genes were present in two orchids, D. officinale and Phalaenopsis equestris,whiletheTM8,FLC,AGL15 and AGL12 subfamilies may be lost. In addition, the four clades of floral organ identity genes in both orchids displayed a conservative and divergent expression pattern. Only three MIKC-type genes were induced by cold stress in D. -

Genome-Wide Study of SBT Genes in Eight Plant Species: Evolution and Expression Outlines for Development and Stress
Genome-wide Study of SBT Genes in Eight Plant Species: Evolution and Expression Outlines for Development and Stress Sulin Wen Shanghai Jiao Tong University https://orcid.org/0000-0002-5994-3439 Guanqun Chen Shanghai Jiao Tong University Tingting Huang Shanghai Jiao Tong University Xiaohui Shen ( [email protected] ) School of Design, Shanghai Jiao Tong University, Shanghai 200240, China. Research article Keywords: Subtilisin-like proteases, Genome-wide study, Evolution, Expression pattern, Arabidopsis, Rice Posted Date: October 8th, 2020 DOI: https://doi.org/10.21203/rs.3.rs-77460/v1 License: This work is licensed under a Creative Commons Attribution 4.0 International License. Read Full License Page 1/22 Abstract Background: Subtilisin-like proteases (or subtilases, SBT), a specic family of serine peptidases, are a very big family in plants. Previous studies showed association of SBTs with environment response and development. However, it is still unclear about the function and features of SBTs. Also, there is little information about SBTs classication or protein function research. Results: The characters of SBTs in eight plant species including Arabidopsis, rice, wheat etc. were analyzed. The analysis mainly contained evolutionary relationship, gene structure, chromosomal location, gene synteny and expression pattern. In short, the evolutionary trend of SBTs in the eight plant species was revealed. The expression pattern showed a scattered distribution of SBTs in diverse plant organs. Conclusions: Our results reveal classication, feature and expression pattern of SBT. Then the results might offer clues for future studies about SBTs and additionally related to their possible function in plants against stress and development. Background Protease is a kind of important enzyme in most living things, which breakdown of proteins into smaller polypeptides or single amino acids. -

PC25 Doc. 32.2
Original language: English PC25 Doc. 32.2 CONVENTION ON INTERNATIONAL TRADE IN ENDANGERED SPECIES OF WILD FAUNA AND FLORA ___________________ Twenty-fifth meeting of the Plants Committee Online, 2-4, 21 and 23 June 2021 Species specific matters Maintenance of the Appendices Orchids checklists APPENDIX-II ORCHID CHECKLIST 1. This document has been submitted by the Scientific Authority for Flora of the United Kingdom of Great Britain and Northern Ireland.* 2. The context of this document pertains to PC24 Com. 8 (Rev. by Sec.). The UK Scientific Authority and the United Nations Environment Programme – World Conservation Monitoring Centre (UNEP-WCMC) were to prepare a checklist for Orchidaceae, presenting Appendix I and Appendix II species separately. a) This was to be undertaken by generating an output for Orchidaceae from the World Checklist of Selected Plant Families. The output includes accepted names, synonyms and country-level distribution information. b) The dataset for Orchidaceae was provided by The World Checklist of Selected Plant Families. The World Checklist of Selected Plant Families has become an international collaborative programme with more than 150 contributors from 22 countries. The main goal of the World Checklist of Selected Plant Families is to provide high quality peer reviewed baseline data on all accepted taxa included in each family. c) To make the review of proposed changes manageable, a comparison was undertaken between the World Checklist of Selected Plant Families output and the current CITES nomenclature standard references for Orchidaceae. 3. The Appendix I Orchid Checklist was adopted at the 18th CITES Conference of the Parties (Switzerland, 2019). This checklist and the proposed checklist were compiled using the same methodology. -

Detection of Somaclonal Variation by Random Amplified Polymorphic DNA Analysis During Micropropagation of Phalaenopsis Bellina (Rchb.F.) Christenson
African Journal of Biotechnology Vol. 9(40), pp. 6632-6639, 4 October, 2010 Available online at http://www.academicjournals.org/AJB DOI: 10.5897/AJB10.714 ISSN 1684–5315 ©2010 Academic Journals Full Length Research Paper Detection of somaclonal variation by random amplified polymorphic DNA analysis during micropropagation of Phalaenopsis bellina (Rchb.f.) Christenson Khoddamzadeh A. A1, Sinniah U. R1*, Kadir M. A2, Kadzimin S. B1, Mahmood M 3 and Sreeramanan S 4 1Department of Crop Science, Faculty of Agriculture, Universiti Putra Malaysia (UPM), 43300, Serdang, Malaysia. 2Department of Agrotechnology, Faculty of Agriculture, Universiti Putra Malaysia (UPM), 43300, Serdang, Malaysia. 3Department of Biochemistry, Faculty of Biotechnology and Biomolecular Sciences, Universiti Putra Malaysia (UPM), 43300, Serdang, Malaysia. 4School of Biological Sciences, Universiti Sains Malaysia (USM), Minden Heights, 11800, Georgetown, Penang, Malaysia. Accepted 23 August, 2010 Phalaenopsis bellina (Rchb.f.) Christenson orchid species are known for their beautiful flower shape, graceful inflorescence and fragrance. Protocorm-like bodies (PLBs) of P. bellina were induced from leaf segments. The PLBs were then subjected to proliferation using ½ strength Murashige and Skoog (MS) media with two subcultures at three months intervals. Twelve decamer random amplified polymorphic DNA (RAPD) primers were used to study somaclonal variation among the mother plant, the initially induced PLBs and proliferated PLBs after 3 and 6 months in culture. Eight out of twelve primers produced 172 bands with 18 polymorphic bands in all the treatments. The amplified products varied between 125 to 8000 bp. Among the primers used, P 16 produced the highest number of bands (29), while primer OPU 10 produced the lowest number (15). -

Index Sorted by Title
Index sorted by Title Volume Issue Year Article Title Author Key Words 31 5 1967 12th Western Orchid Congress Jefferies, George Western Orchid Congress 31 5 1967 12th Western Orchid Congress — Photo Flashes Philpott, R. G. Western Orchid Congress 12th World Orchid Conference ... March 1987, 51 4 1987 Eilau, William World Orchid Conference, Tokyo Tokyo, Japan 13th World Orchid Conference, Auckland, New World Orchid Conference, New 54 2 1990 Eilau, William Zealand Zealand 14th World Orchid Conference, Glascow, 57 3 1993 Hetherington, Ernest World Orchid Conference, scotland Scotland, April 26-May 1, 1993, The 1992 Volume of the Orchid Digest is Dedicated 56 1 1992 in Memoriam to D. George Morel (1926-1973), Hetherington, Ernest history, George Morel The 58 4 1994 1994 Orchid Digest Research Grant Digest Staff 1994 orchid, research, grant 1995 Orchid Digest Dec Dedicated to Herb 59 1 1995 Digest Staff Dedication, Herb Hager Hager 72 2 2008 19th World Orchid Conference Hersch, Helen world orchid conference, 19th 2018 Paphiopedilum Guild and the Second 2018, paphiopedilum guild, second 82 2 2018 International World Slipper Orchid Conference Sorokowsky, David international world slipper orchid, Hilo, Hawaii conference 80 3 2016 22nd World Orchid Conference Pridgeon, Alec 22nd World Orchid Conference 84 4 2020 A Checklist of Phramipedium Species Cervera, Frank checklist, phragmipedium 84 3 2020 A New Color Forma for Vanda curvifolia Koopowitz, Harold vanda, curvifolia, new color form A New Species of Lepanthes (Orchidaceae: Larson, Bruno, Portilla, Jose, Medina 85 2 2021 new species, Lepanthes, Ecuador Pleurothallidinae) from South East Ecuador Hugo A New Species of Pleurothallopsis new species, pleurothallopsis, 82 1 2018 (Epidendreae, Epidendroideae, Orchidaceae): Matthews, Luke M. -
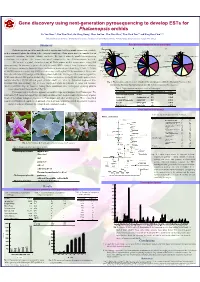
Gene Discovery Using Next-Generation Pyrosequencing To
Gene discovery using next-generation pyrosequencing to develop ESTs for Phalaenopsis orchids Yu-Yun Hsiao1,2, Yun-Wen Chen3, Shi-Ching Huang1, Zhao-Jun Pan1, Wen-Huei Chen2, Wen-Chieh Tsai2,3* and Hong-Hwa Chen1,2,3* 1Department of Life Sciences, 2Orchid Research Center, 3Institute of Tropical Plant Sciences, National Cheng Kung University, Tainan 701, Taiwan Summary Functional annotation of novel transcripts A B Structural Molecule Activity C 2% Orchids represent one of the most diversified angiosperms, but few genomic resources are available Protein Metabolism Signal Transduction ER 1% 1% Response to Abiotic or Biotic 1% Golgi Apparatus Stimulus DNA or RNA binding Plastid Cell Organization And Biogenesis 2% 2% 1% 2% in these non-model plants. In addition to the ecological significance, Phalaenopsis has been considered as an Transcription 1% Nucleic Acid Binding Cytosol 3% 2% 3% Other Intracellular Components Other Molecular Functions Developmental Processes Unknown Biological Processes 4% Other Enzyme Activity 3% Unknown Cellular Components 19% 22% economically important floriculture industry worldwide. We applied massively parallel pyrosequencing 2% 20% Transcription Factor Activity Cell Wall 3% Other Binding 4% 4% Mitochondria Protein Binding 4% technique to explore the transcriptional complexity for Phalaenopsis orchid. Other Biological Processes 4% 4% Other Cellular Components 6% We present a global characterization of Phalaenopsis orchid transcriptome using 454 Kinase Activity Nucleotide Binding 5% 5% pyrosequencing. To maximize sequence diversity, we pooled RNA extracted from ten samples including Other Cytoplasmic Components DNA or RNA binding Other Binding Other Cellular Processes Unknown Molecular Functions 7% 5% 6% different tissues, various developmental stages, and biotic or abiotic stressed plants (Fig. -

Phaldb:A Comprehensive Database for Molecular Mining of the Phalaenopsis Genome, Transcriptome and Mirnome
PhalDB:A comprehensive database for molecular mining of the Phalaenopsis genome, transcriptome and miRNome C.Y. Lee1, K.K. Viswanath1, J.Z. Huang1, C.P. Lee2, C.P. Lin2,4, T.C. Cheng1, B. C. Chang2,3, S.W. Chin1, and F.C. Chen1 1 Department of Plant Industry, National Pingtung University of Science and Technology, Pingtung, Taiwan 2 Yourgene Bioscience, Shu-Lin District, New Taipei City, Taiwan 3 Faculty of Veterinary Science, The University of Melbourne, Parkville, Victoria, Australia 4 Department of Biotechnology, School of Health Technology, Ming Chuan University, Gui Shan District, Taoyuan, Taiwan Corresponding author: F.C. Chen E-mail: [email protected] Genet. Mol. Res. 17 (4): gmr18051 Received June 06, 2018 Accepted July 26, 2018 Published October 05, 2018 DOI http://dx.doi.org/10.4238/gmr18051 ABSTRACT. The orchid family Orchidaceae is one of the largest families of angiosperms; it includes approximately 35,000 species, displays a wide range of unique flower shapes and is a valued ornamental crop. Among the various species of orchids, the Phalaenopsis genus is the most widely commercialized among blooming potted plants. Our aim was to provide in-depth sequence information for the various parts of flowers, different time treatments of the protocorm-like bodies and temperature treated shortened stems for flowering pathway regulation, in addition to adding to the available data on normal and peloric mutants. We also obtained transcriptome data from shortened stems under cool temperature for spike induction. The deep sequenced, assembled and annotated information was integrated and built into a database that we named PhalDB. -

Hasil Seleksi Abstrak Untuk Presentasi Oral Pada
HASIL SELEKSI ABSTRAK UNTUK PRESENTASI ORAL PADA SEMINAR NASIONAL HORTIKULTURA 2010 Diilaksanakan oleh Perhimpunan Hortikultura Indonesia (PERHORTI) bekerjasama dengan Pusat Penelitian dan Pengembangan Hortikultura, Universitas Udayana Seminar diselenggarakan pada tanggal 25-26 November 2010 di Kampus Sudirman, Universitas Udayana No Nama Teregistrasi Intitusi dan Alamat Alamat E-mail Judul Paper / Artikel Deden Derajat Matra, PT. Santini Mitra Amanah Pengelompokan Populasi Manggis 1 Roedhy Poerwanto, Edi (Gunung Geulis Orchid [email protected] Berdasarkan Karakter Fenotipe di Jawa Santosa House) dengan Analisis Multivariat Fakultas Pertanian Unversitas Karakterisasi dan Indentifikasi Klon-klon [email protected] 2 Hidayat Tanjungpura, Pontianak. Jl. Lidah Buaya Hasil Pemuliaan Secara Mutasi A. Yani, Pontianak, 78124 dengan Penggunaan EMS A. Aina Roosda, A. Ismail, Program Sarjana Studi Evaluasi Dan Seleksi F1 Ubi Jalar (Ipomoea 3 W. Chandria, A. Pamuliaan Tanaman Fakultas [email protected] batatas (L.) Lam) ex Jepang Outcrossing Karuniawan Pertanian UNPAD Berdasarkan Karakter Hasil Di Jatinangor Anggie, E.P., Rineza K. Genetic parameters of baby corn DR Unpad Limindo, Winny, W., T. hybrids in different plant densities Lab. Pemuliaan tanaman, dediruswandi2000@yahoo. 4 Mutiarawati, S R . (Parameter genetik karakter jagung semi Universitas Padjadjaran com Ruswandi , and D. pada hibrida DR dalam beberapa kerapatan Ruswandi tanaman) BPTP Kalimantan Barat Arry Supriyanto, M. Jl. Budi Utomo No. 45 Siantan Karateristik Buah Langsat (Lansium 5 Zuhran, Tommy Purba, dan [email protected]. Hulu Pontianak domesticum) Punggur di Kalimantan Barat. Titiek Kartinaty Balai Pengkajian Teknologi Baswarsiati, D. Rahmawati, Sistem Perbenihan dan Produksi Benih 6 Pertanian Jawa Timur [email protected] S.Purnomo, Abu Bawang Merah "Sumenep" . Jl Raya Karangploso Km-4, Malang Penampilan Fenotipik, Heritabilitas dan Chindy Ulima Z., Agung Respons Seleksi Karakter Komponen Hasil Program Studi Pemuliaan chindy_u5z4n3tta@yahoo.