Iiiiiiih 3 1293 03046 3 9
Total Page:16
File Type:pdf, Size:1020Kb

Load more
Recommended publications
-

WO 2015/179628 Al 26 November 2015 (26.11.2015) P O P C T
(12) INTERNATIONAL APPLICATION PUBLISHED UNDER THE PATENT COOPERATION TREATY (PCT) (19) World Intellectual Property Organization International Bureau (10) International Publication Number (43) International Publication Date WO 2015/179628 Al 26 November 2015 (26.11.2015) P O P C T (51) International Patent Classification: (81) Designated States (unless otherwise indicated, for every C08F 210/16 (2006.01) kind of national protection available): AE, AG, AL, AM, AO, AT, AU, AZ, BA, BB, BG, BH, BN, BR, BW, BY, (21) International Application Number: BZ, CA, CH, CL, CN, CO, CR, CU, CZ, DE, DK, DM, PCT/US20 15/03 1952 DO, DZ, EC, EE, EG, ES, FI, GB, GD, GE, GH, GM, GT, (22) International Filing Date: HN, HR, HU, ID, IL, IN, IR, IS, JP, KE, KG, KN, KP, KR, 2 1 May 2015 (21 .05.2015) KZ, LA, LC, LK, LR, LS, LU, LY, MA, MD, ME, MG, MK, MN, MW, MX, MY, MZ, NA, NG, NI, NO, NZ, OM, (25) Filing Language: English PA, PE, PG, PH, PL, PT, QA, RO, RS, RU, RW, SA, SC, (26) Publication Language: English SD, SE, SG, SK, SL, SM, ST, SV, SY, TH, TJ, TM, TN, TR, TT, TZ, UA, UG, US, UZ, VC, VN, ZA, ZM, ZW. (30) Priority Data: 14/284,689 22 May 2014 (22.05.2014) US (84) Designated States (unless otherwise indicated, for every kind of regional protection available): ARIPO (BW, GH, (71) Applicant: CHEVRON PHILLIPS CHEMICAL COM¬ GM, KE, LR, LS, MW, MZ, NA, RW, SD, SL, ST, SZ, PANY LP [US/US]; 10001 Six Pines Drive, The Wood TZ, UG, ZM, ZW), Eurasian (AM, AZ, BY, KG, KZ, RU, lands, TX 77380 (US). -

CF 7.4 V2.Indd
2007 VOLUME 7 NUMBER 4 Product Directory Grignard and Organozinc Reagents RIEKE® HIGHLY REACTIVE METALS GRIGNARD REAGENTS ORGANOZINC HALIDES DIALKYLMAGNESIUM AND DIALKYZINC REAGENTS 2-Pyridylzinc bromide: a shelf-stable 2-pyridyl anion equivalent; an important motif in many pharmacologically active molecules. sigma-aldrich.com 2 Table of Contents Sigma-Aldrich is committed to providing the most extensive portfolio of high-quality Grignard, organozinc, and other organometallic reagents, and we continually expand our product listing. Within each section of this directory, products are listed by increasing carbon content. Rieke® Highly Reactive Metals If viewing the electronic version simply select Grignard Reagents a category to jump to that section or activate Alkyl Alkenyl Alkynyl Aryl Heteroaryl your Adobe Bookmarks. You may also search by name, product number, molecular formula, Organozinc Halides or CAS registry number simply by using the “find” feature in Adobe (Ctrl+F in Windows or Alkyl Alkenyl Aryl Heteroaryl Introduction Command+F in a Mac environment). Dialkylmagnesium and Dialkylzinc Reagents If you are unable to find a reagent for your research “Please Bother Us” at [email protected], or contact your local Sigma-Aldrich office (see back cover). Foreword Reuben D. Rieke President and CEO, Rieke Metals, Inc. Professor Emeritus, University of Nebraska Lincoln, NE In the last 35 years, considerable research has been done in the area of generating reactive metals that can be used to synthesize novel organometallic reagents. In 1972, we reported a general approach for preparing highly reactive metal powders, relying on the reduction of metal salts with alkali metals in ethereal or hydrocarbon solvents. -

Chemical Name Federal P Code CAS Registry Number Acutely
Acutely / Extremely Hazardous Waste List Federal P CAS Registry Acutely / Extremely Chemical Name Code Number Hazardous 4,7-Methano-1H-indene, 1,4,5,6,7,8,8-heptachloro-3a,4,7,7a-tetrahydro- P059 76-44-8 Acutely Hazardous 6,9-Methano-2,4,3-benzodioxathiepin, 6,7,8,9,10,10- hexachloro-1,5,5a,6,9,9a-hexahydro-, 3-oxide P050 115-29-7 Acutely Hazardous Methanimidamide, N,N-dimethyl-N'-[2-methyl-4-[[(methylamino)carbonyl]oxy]phenyl]- P197 17702-57-7 Acutely Hazardous 1-(o-Chlorophenyl)thiourea P026 5344-82-1 Acutely Hazardous 1-(o-Chlorophenyl)thiourea 5344-82-1 Extremely Hazardous 1,1,1-Trichloro-2, -bis(p-methoxyphenyl)ethane Extremely Hazardous 1,1a,2,2,3,3a,4,5,5,5a,5b,6-Dodecachlorooctahydro-1,3,4-metheno-1H-cyclobuta (cd) pentalene, Dechlorane Extremely Hazardous 1,1a,3,3a,4,5,5,5a,5b,6-Decachloro--octahydro-1,2,4-metheno-2H-cyclobuta (cd) pentalen-2- one, chlorecone Extremely Hazardous 1,1-Dimethylhydrazine 57-14-7 Extremely Hazardous 1,2,3,4,10,10-Hexachloro-6,7-epoxy-1,4,4,4a,5,6,7,8,8a-octahydro-1,4-endo-endo-5,8- dimethanonaph-thalene Extremely Hazardous 1,2,3-Propanetriol, trinitrate P081 55-63-0 Acutely Hazardous 1,2,3-Propanetriol, trinitrate 55-63-0 Extremely Hazardous 1,2,4,5,6,7,8,8-Octachloro-4,7-methano-3a,4,7,7a-tetra- hydro- indane Extremely Hazardous 1,2-Benzenediol, 4-[1-hydroxy-2-(methylamino)ethyl]- 51-43-4 Extremely Hazardous 1,2-Benzenediol, 4-[1-hydroxy-2-(methylamino)ethyl]-, P042 51-43-4 Acutely Hazardous 1,2-Dibromo-3-chloropropane 96-12-8 Extremely Hazardous 1,2-Propylenimine P067 75-55-8 Acutely Hazardous 1,2-Propylenimine 75-55-8 Extremely Hazardous 1,3,4,5,6,7,8,8-Octachloro-1,3,3a,4,7,7a-hexahydro-4,7-methanoisobenzofuran Extremely Hazardous 1,3-Dithiolane-2-carboxaldehyde, 2,4-dimethyl-, O- [(methylamino)-carbonyl]oxime 26419-73-8 Extremely Hazardous 1,3-Dithiolane-2-carboxaldehyde, 2,4-dimethyl-, O- [(methylamino)-carbonyl]oxime. -

Grignard Reagents and Silanes
GRIGNARD REAGENTS AND SILANES By B. Arkles REPRINTED FROM HANDBOOK OF GRIGNARD REAGENTS by G. Silverman and P. Rakita Pages 667-675 Marcel Dekker, 1996 Gelest, Inc. 612 William Leigh Drive Tullytown, Pa. 19007-6308 Phone: [215] 547-1015 Fax: [215] 547-2484 Grignard Reagents and Silanes 32 Grignard Reagents and Silanes BARRY ARKLES Gelest Inc., Tullytown, Pennsylvania I. INTRODUCTION This review considers two aspects of the interaction of Grignards with silanes. First, focusing on technologies that are still viable within the context of current organosilane and silicone technology, guidelines are provided for silicon-carbon bond formation using Grignard chemistry. Second, the use of silane-blocking agents and their stability in the presence of Grignard reagents employed in organic synthesis is discussed. II. FORMATION OF THE SILICON-CARBON BOND A. Background The genesis of current silane and silicone technology traces back to the Grignard reaction. The first practical synthesis of organosilanes was accomplished by F. Stanley Kipping in 1904 by the Grignard reaction for the formation of the silicon-carbon bond [1]. In an effort totaling 57 papers, he created the basis of modern organosilane chemistry. The development of silicones by Frank Hyde at Corning was based on the hydrolysis of Grignard-derived organosilanes [2]. Dow Corning, the largest manufacturer of silanes and silicones, was formed as a joint venture between Corning Glass, which had silicone product technology, and Dow, which had magnesium and Grignard technology, during World War II. In excess of 10,000 silicon compounds have been synthesized by Grignard reactions. Ironically, despite the versatility of Grignard chemistry for the formation of silicon-carbon bonds, its use in current silane and silicone technology has been supplanted by more efficient and selective processes for the formation of the silicon-carbon bond, notably by the direct process and hydrosilylation reactions. -

Acutely / Extremely Hazardous Waste List
Acutely / Extremely Hazardous Waste List Federal P CAS Registry Acutely / Extremely Chemical Name Code Number Hazardous 4,7-Methano-1H-indene, 1,4,5,6,7,8,8-heptachloro-3a,4,7,7a-tetrahydro- P059 76-44-8 Acutely Hazardous 6,9-Methano-2,4,3-benzodioxathiepin, 6,7,8,9,10,10- hexachloro-1,5,5a,6,9,9a-hexahydro-, 3-oxide P050 115-29-7 Acutely Hazardous Methanimidamide, N,N-dimethyl-N'-[2-methyl-4-[[(methylamino)carbonyl]oxy]phenyl]- P197 17702-57-7 Acutely Hazardous 1-(o-Chlorophenyl)thiourea P026 5344-82-1 Acutely Hazardous 1-(o-Chlorophenyl)thiourea 5344-82-1 Extemely Hazardous 1,1,1-Trichloro-2, -bis(p-methoxyphenyl)ethane Extemely Hazardous 1,1a,2,2,3,3a,4,5,5,5a,5b,6-Dodecachlorooctahydro-1,3,4-metheno-1H-cyclobuta (cd) pentalene, Dechlorane Extemely Hazardous 1,1a,3,3a,4,5,5,5a,5b,6-Decachloro--octahydro-1,2,4-metheno-2H-cyclobuta (cd) pentalen-2- one, chlorecone Extemely Hazardous 1,1-Dimethylhydrazine 57-14-7 Extemely Hazardous 1,2,3,4,10,10-Hexachloro-6,7-epoxy-1,4,4,4a,5,6,7,8,8a-octahydro-1,4-endo-endo-5,8- dimethanonaph-thalene Extemely Hazardous 1,2,3-Propanetriol, trinitrate P081 55-63-0 Acutely Hazardous 1,2,3-Propanetriol, trinitrate 55-63-0 Extemely Hazardous 1,2,4,5,6,7,8,8-Octachloro-4,7-methano-3a,4,7,7a-tetra- hydro- indane Extemely Hazardous 1,2-Benzenediol, 4-[1-hydroxy-2-(methylamino)ethyl]- 51-43-4 Extemely Hazardous 1,2-Benzenediol, 4-[1-hydroxy-2-(methylamino)ethyl]-, P042 51-43-4 Acutely Hazardous 1,2-Dibromo-3-chloropropane 96-12-8 Extemely Hazardous 1,2-Propylenimine P067 75-55-8 Acutely Hazardous 1,2-Propylenimine 75-55-8 Extemely Hazardous 1,3,4,5,6,7,8,8-Octachloro-1,3,3a,4,7,7a-hexahydro-4,7-methanoisobenzofuran Extemely Hazardous 1,3-Dithiolane-2-carboxaldehyde, 2,4-dimethyl-, O- [(methylamino)-carbonyl]oxime 26419-73-8 Extemely Hazardous 1,3-Dithiolane-2-carboxaldehyde, 2,4-dimethyl-, O- [(methylamino)-carbonyl]oxime. -

Acroseal Packaging Your Solution for Air- and Moisture- Sensitive Reagents
AcroSeal Packaging Your solution for air- and moisture- sensitive reagents Extra dry solvents Deuterated solvents Organometallic compounds Reagents in solution Organics Introduction Since the launch of AcroSealTM packaging we have introduced a new septum, which helps preserve product quality for longer. In addition, our AcroSeal portfolio has been expanded to include a broad range of solvents, organometallics, reagents in solution and organic compounds. In this brochure we have categorized our products under chemical families to make it easier to locate the product you need. Introduction Page no. AcroSeal packaging highlights 3 AcroSeal packaging performance 4 New 25mL AcroSeal packaging 4 Solvents Extra dry solvents 5-7 Solvents for biochemistry 7 Deuterated solvents 7 Organometallics Grignard reagents 8-10 Organoaluminiums 11 Organolithiums 11 Organosodiums 12 Organotins 12 Organozincs 12 Reagents in solution Amines 13 Boranes 13 Halides 14-15 Hydrides 15 Oxides 16 Silanes 16 Other reagents in solution 17 Organics Aldehydes 18 Amines 18 Epoxides 18 Halides 19 Phosphines 19 Silanes 19 Other organics 20 How to use AcroSeal packaging 21 Alphabetical index 22-23 2 Introduction AcroSeal packaging: drier reagents for longer When using air- and moisture-sensitive solvents and reagents, it is essential that these products are not only as dry as possible when you first use them, but they should remain dry in storage as well. Through the innovative quadrant-style screw cap and specially designed septum, AcroSeal packaging ensures that you have access to high-quality and low-moisture products every use, guaranteeing improved yield and consistency of your research experiments while reducing chemical waste. AcroSeal packaging highlights New septum developed from a polymeric elastomer with an inert fluoropolymer-coated surface, preserves product quality for longer with better re-seal around needle punctures. -

Electrodeposition of Magnesium and Beryllium from Organic Baths
NASA CR-111850 ELECTRODEPOSITION OF MAGNESIUM AND BERYLLIUM FROM ORGANIC BATHS by Abner Brenner and John L. Sligh Prepared under Purchase Request L-37,566 by NATIONAL BUREAU OF STANDARDS Washington, DC 20234 for NATIONAL URONAUTICS AND SPACE ADMINISTWTION ELECTRODEPOSITION OF MAGNESIUM AND BERYLLIUM FROM 0,RGANIC BATHS Abner Brenner and John Le Sligh National Bureau of Standards, Washington, D. C. 20234 ABSTRACT Solutions that are feasible for electrodepositing magnesium were prepared from Grignard reagents with the addition of boranes in ether solutions. A typical bath consisted of methylmagnesium chloride, 3-molar9 (dissolved in tetrahydrofuran) and l-molar in triethylboron. The deposits were white, ductile and contained at least 99% of Mg. Cathode and anode current efficiencies were 100%. A similar beryllium plating solution, prepared with beryllium dimethyl and decaborane, yielded dark grey, coherent deposits containing 85% to 90% beryllium. KEY WORDS: Magnesium, electrodeposition of; Beryllium, electrodeposition of; nonaqueous plating baths; Grignard reagents; Beryllium dimethyl; Beryllium azide; Beryllium thiocyanate, decaborane; triethylboron; Organic plating baths. ELECTRODEPOSITION OF MAGNESIUM AND BERYLLIUM FROM ORGANIC BATHS Abner Brenner and John Le Sligh National Bureau of Standards, Washington, D. C. 20234 I e INTRODUCTION The chief interest in electrodepositing the light structural metals, beryllium, magnesium, and aluminum is for the purpose of electroforming light-weight engineering components of complex shape or precise dimen- sions that are needed in satellites. These metals are too active to be electrodeposited from aqueous solutions, hence, organic baths must be used. Of the three metals, aluminum is the only one that has been suc- cessfully electrodeposited and used in pilot-scale investigations. -

Asymmetric Hydrogenation Catalysts to Conductive Polymers. Mark Steven Erickson Louisiana State University and Agricultural & Mechanical College
Louisiana State University LSU Digital Commons LSU Historical Dissertations and Theses Graduate School 1992 Synthesis of Novel Metallocenes: Asymmetric Hydrogenation Catalysts to Conductive Polymers. Mark Steven Erickson Louisiana State University and Agricultural & Mechanical College Follow this and additional works at: https://digitalcommons.lsu.edu/gradschool_disstheses Recommended Citation Erickson, Mark Steven, "Synthesis of Novel Metallocenes: Asymmetric Hydrogenation Catalysts to Conductive Polymers." (1992). LSU Historical Dissertations and Theses. 5431. https://digitalcommons.lsu.edu/gradschool_disstheses/5431 This Dissertation is brought to you for free and open access by the Graduate School at LSU Digital Commons. It has been accepted for inclusion in LSU Historical Dissertations and Theses by an authorized administrator of LSU Digital Commons. For more information, please contact [email protected]. INFORMATION TO USERS This manuscript has been reproduced from the microfilm master. UMI films the text directly from the original or copy submitted. Thus, some thesis and dissertation copies are in typewriter face, while others may be from any type of computer printer. The quality of this reproduction is dependent upon the quality of the copy submitted. Broken or indistinct print, colored or poor quality illustrations and photographs, print bleedthrough, substandard margins, and improper alignment can adversely affect reproduction. In the unlikely event that the author did not send UMI a complete manuscript and there are missing pages, these will be noted. Also, if unauthorized copyright material had to be removed, a note will indicate the deletion. Oversize materials (e.g., maps, drawings, charts) are reproduced by sectioning the original, beginning at the upper left-hand corner and continuing from left to right in equal sections with small overlaps. -

(12) United States Patent (10) Patent No.: US 6,313,230 B1 Tsai Et Al
USOO631323OB1 (12) United States Patent (10) Patent No.: US 6,313,230 B1 Tsai et al. (45) Date of Patent: Nov. 6, 2001 (54) CATALYST COMPOSITION FOR 6,228,952 5/2001 Viola et al. .......................... 525/338 HYDROGENATION OF CONJUGATED * cited b DIENE BASED SYNTHETIC RUBBERS cited by examiner Primary Examiner Bernard Lipman (75) Inventors: Jing-Cherng Tsai, Hsinchu; (74) Attorney, Agent, or Firm-Sughrue, Mion, Zinn, Wen-Sheng Chang, Miaoli; Yu-Shan Macpeak & Seas, PLLC Chao, Hsinchu, Chih-Nan Chu, s Tainan; Chen-Pao Huang, Tainan; (57) ABSTRACT Hung-Yang Hsiao, Tainan,ainan, all of (TW)(TW A process for hydrogenation of conjugated diene polymers (73) Assignees: Industrial Technology Research comprises hydrogenating said polymer in the presence of Institute, Hsinchu; Chi Mei hydrogen and a hydrogenation catalyst composition com Corporation, Tainan County, both of pr1SIng: (TW) (a) at least one titanium compound represented by the following formula (a): (*) Notice: Subject to any disclaimer, the term of this patent is extended or adjusted under 35 (a) U.S.C. 154(b) by 0 days. R1 (21) Appl. No.: 09/400,544 (Cp "y (22) Filed: Sep. 21, 1999 (51) Int. C.7 - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - CO8F 8/04 wherein R and R°, which may be the same or different, (52) U.S. Cl. ................... 525/338; 525/332.8; 525/332.9; represent a halogen atom, an alkyl group, an aryl 525/333.2; 525/339 group, an arakyl group, a cycloalkyl group, an ary (58) Field of Search ...................................... 525/338,339 loxy group, an alkoxy group or a carbonyl group, and Cp* represents a cyclopentadienyl group or a (56) References Cited derivative having the formula of CRs, and Rs. -

Hazardous Waste List (California Code of Regulations, Title 22 Section 66261.126)
Hazardous Waste List (California Code of Regulations, Title 22 Section 66261.126) Appendix X - List of Chemical Names and Common Names for Hazardous Wastes and Hazardous Materials (a) This subdivision sets forth a list of chemicals which create a presumption that a waste is a hazardous waste. If a waste consists of or contains a chemical listed in this subdivision, the waste is presumed to be a hazardous waste Environmental Regulations of CALIFORNIA unless it is determined that the waste is not a hazardous waste pursuant to the procedures set forth in section 66262.11. The hazardous characteristics which serve as a basis for listing the chemicals are indicated in the list as follows: (X) toxic (C) corrosive (I) ignitable (R) reactive * =Extremely Hazardous A chemical denoted with an asterisk is presumed to be an extremely hazardous waste unless it does not exhibit any of the criteria set forth in section 66261.110 and section 66261.113. Trademark chemical names are indicated by all capital letters. 1. Acetaldehyde (X,I) 2. Acetic acid (X,C,I) 3. Acetone, Propanone (I) 4. *Acetone cyanohydrin (X) 5. Acetonitrile (X,I) 6. *2-Acetylaminofluorene, 2-AAF (X) 7. Acetyl benzoyl peroxide (X,I,R) 8. *Acetyl chloride (X,C,R) 9. Acetyl peroxide (X,I,R) 10. Acridine (X) 11. *Acrolein, Aqualin (X,I) 12. *Acrylonitrile (X,I) 13. *Adiponitrile (X) 14. *Aldrin; 1,2,3,4,10,10-Hexachloro-1,4,4a,5,8,8a-hexahydro-1,4,5,8-endo-exodimethanonaphthlene (X) 15. *Alkyl aluminum chloride (C,I,R) 16. *Alkyl aluminum compounds (C,I,R) 17. -
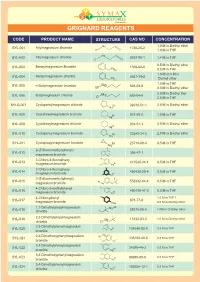
Grignard Reagents
GRIGNARD REAGENTS CODE PRODUCT NAME STRUCTURE CAS NO CONCENTRATION Mg 1.0 M in Diethyl ether SYL-001 Allylmagnesium Bromide 1730-25-2 Br 1.0 M in THF Mg SYL-002 Allylmagnesium chloride 2622-05-1 1.4 M in THF Cl 0.5 M in Diethyl ether Benzylmagnesium Bromide SYL-003 Mg 1589-82-8 Br 0.5 M in THF 1.0 M /2.0 M in SYL-004 Benzylmagnesium chloride Mg 6921-34-2 Cl Diethyl ether 1.0 M in THF SYL-005 n-Butylmagnesium bromide Mg 693-03-8 Br 3.0 M in Diethyl ether 2.0 M in Diethyl ther SYL-006 Butylmagnesium chloride M g 693-04-9 Cl 2.0 M in THF SYLG-007 Cyclopentylmagnesium chloride Cl 32916-51-1 2.0 M in Diethyl ether Mg SYL-008 Cyclohexylmagnesium bromide Br 931-50-0 1.0 M in THF Mg Cyclohexylmagnesium chloride 2.0 M in Diethyl ether SYL-009 Cl 931-51-1 Mg SYL-010 Cyclopentylmagnesium bromide Br 33240-34-5 2.0 M in Diethyl ether Mg SYL-011 Cyclopropylmagnesium bromide Br 23719-80-4 0.5 M in THF Mg F F 2-(Trifluoromethyl)phenyl) F SYL-012 Br 395-47-1 magnesium bromide Mg 3-Chloro-4-fluoropheny F SYL-013 Br 413589-34-1 0.5 M in THF lmagnesium bromide Cl Mg Cl 3-Chloro-5-fluoropheny SYL-014 480438-50-4 0.5 M in THF Br lmagnesium bromide F Mg 3-(Trifluoromethyl)phenyl) F F SYL-015 Br 552332-04-4 0.5 M in THF magnesium bromide Mg O F 4-Chloro-2-methylphenyl Cl SYL-016 Br 480438-47-9 0.5 M in THF magnesium bromide Mg 4-Chloro-phenyl CI 1.0 M in THF / SYL-017 873-77-8 magnesium bromide MgBr 2.0 M in Diethyl ether 1,1-Dimethylpropylmagnesium Cl SYL-018 Mg 28276-08-6 1.0M in Di Ethyl ether chloride 2,2-Dimethylpropylmagnesium SYL-019 Mg 13132-23-5 1.0 -

Hazardous Substances and Waste Dangerous Goods Regulations
1 HAZARDOUS SUBSTANCES AND WASTE DANGEROUS GOODS E-10.2 REG 3 The Hazardous Substances and Waste Dangerous Goods Regulations being Chapter E-10.2 Reg 3 (effective April 1, 1989) as amended by Saskatchewan Regulations 25/92, 107/92, 28/94, 3/95 and 63/2000. NOTE: This consolidation is not official. Amendments have been incorporated for convenience of reference and the original statutes and regulations should be consulted for all purposes of interpretation and application of the law. In order to preserve the integrity of the original statutes and regulations, errors that may have appeared are reproduced in this consolidation. 2 HAZARDOUS SUBSTANCES AND E-10.2 REG 3 WASTE DANGEROUS GOODS Table of Contents 1 Title 11 Decision to grant approval INTERPRETATION 12 Approval not assignable, exception 2 Interpretation 13 Duties of operator, owner HAZARDOUS SUBSTANCES 14 Prohibition re storage in above-ground tanks 3 Designation of hazardous substances 15 Prohibition re storage in underground tanks 16 Prohibition re storage in certain containers or DESIGNATION AS HAZARDOUS WASTES stockpiles 3.1 Designation of waste dangerous goods 17 Decommissioning as hazardous wastes TRANSFERAL OF WASTE DANGEROUS GOODS CHARACTERIZATION OF SUBSTANCES 18 Transferal of waste dangerous goods 4 Characteristics of certain hazardous substances EXEMPTION FROM REQUIREMENTS Appendix A 5 General Exemptions INDUSTRIAL HAZARDOUS SUBSTANCES 6 Underground storage facilities Appendix B 7 Above-ground storage facilities ACUTE HAZARDOUS SUBSTANCES 8 Storage in small containers Appendix C APPROVAL TO STORE ENVIRONMENTAL PERSISTENT OR 9 Approval to store CHRONIC HAZARDOUS SUBSTANCES APPROVAL TO CONSTRUCT Appendix D 10 Approval to construct WASTE DANGEROUS GOODS 3 HAZARDOUS SUBSTANCES AND WASTE DANGEROUS GOODS E-10.2 REG 3 CHAPTER E-10.2 REG 3 The Environmental Management and Protection Act Title 1 These regulations may be cited as The Hazardous Substances and Waste Dangerous Goods Regulations.