Insulin Resistance Uncoupled from Dyslipidemia Due to C- Terminal PIK3R1 Mutations
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Deregulated Gene Expression Pathways in Myelodysplastic Syndrome Hematopoietic Stem Cells
Leukemia (2010) 24, 756–764 & 2010 Macmillan Publishers Limited All rights reserved 0887-6924/10 $32.00 www.nature.com/leu ORIGINAL ARTICLE Deregulated gene expression pathways in myelodysplastic syndrome hematopoietic stem cells A Pellagatti1, M Cazzola2, A Giagounidis3, J Perry1, L Malcovati2, MG Della Porta2,MJa¨dersten4, S Killick5, A Verma6, CJ Norbury7, E Hellstro¨m-Lindberg4, JS Wainscoat1 and J Boultwood1 1LRF Molecular Haematology Unit, NDCLS, John Radcliffe Hospital, Oxford, UK; 2Department of Hematology Oncology, University of Pavia Medical School, Fondazione IRCCS Policlinico San Matteo, Pavia, Italy; 3Medizinische Klinik II, St Johannes Hospital, Duisburg, Germany; 4Division of Hematology, Department of Medicine, Karolinska Institutet, Stockholm, Sweden; 5Department of Haematology, Royal Bournemouth Hospital, Bournemouth, UK; 6Albert Einstein College of Medicine, Bronx, NY, USA and 7Sir William Dunn School of Pathology, University of Oxford, Oxford, UK To gain insight into the molecular pathogenesis of the the World Health Organization.6,7 Patients with refractory myelodysplastic syndromes (MDS), we performed global gene anemia (RA) with or without ringed sideroblasts, according to expression profiling and pathway analysis on the hemato- poietic stem cells (HSC) of 183 MDS patients as compared with the the French–American–British classification, were subdivided HSC of 17 healthy controls. The most significantly deregulated based on the presence or absence of multilineage dysplasia. In pathways in MDS include interferon signaling, thrombopoietin addition, patients with RA with excess blasts (RAEB) were signaling and the Wnt pathways. Among the most signifi- subdivided into two categories, RAEB1 and RAEB2, based on the cantly deregulated gene pathways in early MDS are immuno- percentage of bone marrow blasts. -

Phosphorylation of Plcg1 by Epha2 Receptor Tyrosine Kinase Promotes Tumor Growth in Lung Cancer Wenqiang Song1,2, Laura C
Published OnlineFirst August 4, 2020; DOI: 10.1158/1541-7786.MCR-20-0075 MOLECULAR CANCER RESEARCH | SIGNAL TRANSDUCTION AND FUNCTIONAL IMAGING Phosphorylation of PLCg1 by EphA2 Receptor Tyrosine Kinase Promotes Tumor Growth in Lung Cancer Wenqiang Song1,2, Laura C. Kim3, Wei Han4, Yuan Hou5, Deanna N. Edwards1, Shan Wang1, Timothy S. Blackwell2,4,6, Feixiong Cheng5,7,8, Dana M. Brantley-Sieders1,9, and Jin Chen1,2,3,6,9 ABSTRACT ◥ EphA2 receptor tyrosine kinase (RTK) is often expressed at EphA2 decreased phosphorylation of PLCg1andlossofPLCg1 high levels in cancer and has been shown to regulate tumor inhibited tumor cell growth in vitro.KnockoutofPLCg1by growth and metastasis across multiple tumor types, including CRISPR-mediated genome editing also impaired tumor growth non–small cell lung cancer. A number of signaling pathways in a KrasG12D-p53-Lkb1 murine lung tumor model. Collectively, downstream of EphA2 RTK have been identified; however, these data show that the EphA2-PLCg1 signaling axis promotes mechanisms of EphA2 proximal downstream signals are less tumor growth of lung cancer and provides rationale for disrup- well characterized. In this study, we used a yeast-two-hybrid tion of this signaling axis as a potential therapeutic option. screen to identify phospholipase C gamma 1 (PLCg1) as a novel EphA2 interactor. EphA2 interacts with PLCg1andthekinase Implications: The EphA2-PLCG1 signaling axis promotes tumor activity of EphA2 was required for phosphorylation of PLCg1. In growth of non–small cell lung cancer and can potentially be targeted human lung cancer cells, genetic or pharmacologic inhibition of as a therapeutic option. Introduction inhibitor–resistant tumor cells (5). -

MERTK-Mediated Signaling in the Retinal Pigment Epithelium: Insights Into the Mechanism of RPE Phagocytosis
MERTK-mediated Signaling in the Retinal Pigment Epithelium: Insights into the Mechanism of RPE Phagocytosis by Shameka J. Shelby A dissertation submitted in partial fulfillment of the requirements for the degree of Doctor of Philosophy (Biological Chemistry) in The University of Michigan 2012 Doctoral Committee: Professor Debra A. Thompson, Chair Professor Christin Carter-Su Professor Bret A. Hughes Professor Benjamin L. Margolis Assistant Professor Hisashi Umemori © Shameka J. Shelby 2012 To my two favorite men, Juan and Zani for your patience, love, and support. ii Acknowledgements I would like to acknowledge all of the people who helped me to accomplish my goals. My mentor, Dr. Debra Thompson, has been instrumental in my success as a scientist. Her motherly guidance has helped to mold me into the person and scientist I am today. I’m overly grateful for her tireless efforts and patience during the course of my graduate career. I wouldn’t have even considered graduate school without the guidance of my undergraduate advisors, Drs. Marion Carroll and Guangdi Wang, to whom I am also grateful. Also, my favorite high school teacher Mrs. Jenkins incited my passion for biology and encouraged me to follow my aspirations. I am also grateful for the invaluable input and criticisms of my committee members, Drs. Bret Hughes, Christin Carter-Su, Hisashi Umemori, and Benjamin Margolis. I would especially like to thank Dr. Hughes, Dr. Carter-Su, and Dr. Margolis for reagents that were instrumental in the completion of my experiments. Ben literally saved my life with the plethora of antibodies he provided. Reagents and clones provided by Drs. -

Anti-VEGFR2 Phospho (Tyr1214) Antibody (ARG51588)
Product datasheet [email protected] ARG51588 Package: 100 μl, 50 μl anti-VEGFR2 phospho (Tyr1214) antibody Store at: -20°C Summary Product Description Rabbit Polyclonal antibody recognizes VEGFR2 phospho (Tyr1214) Tested Reactivity Hu, Ms Tested Application ICC/IF, IHC-P, WB Host Rabbit Clonality Polyclonal Isotype IgG Target Name VEGFR2 Antigen Species Human Immunogen Peptide sequence around phosphorylation site of tyrosine 1214 (F-H-Y(p)-D-N) derived from Human VEGFR2. Conjugation Un-conjugated Alternate Names FLK1; VEGFR; CD antigen CD309; FLK-1; Fetal liver kinase 1; VEGFR2; Vascular endothelial growth factor receptor 2; VEGFR-2; CD309; Kinase insert domain receptor; EC 2.7.10.1; Protein-tyrosine kinase receptor flk-1; KDR Application Instructions Application table Application Dilution ICC/IF 1:100 - 1:200 IHC-P 1:50 - 1:100 WB 1:500 - 1:1000 Application Note * The dilutions indicate recommended starting dilutions and the optimal dilutions or concentrations should be determined by the scientist. Calculated Mw 152 kDa Properties Form Liquid Purification Antibodies were produced by immunizing rabbits with KLH-conjugated synthetic phosphopeptide. Antibodies were purified by affinity-chromatography using epitope-specific phosphopeptide. In addition, non-phospho specific antibodies were removed by chromatogramphy using non- phosphopeptide. Buffer PBS (without Mg2+ and Ca2+, pH 7.4), 150mM NaCl, 0.02% Sodium azide and 50% Glycerol. Preservative 0.02% Sodium azide Stabilizer 50% Glycerol www.arigobio.com 1/3 Concentration 1 mg/ml Storage instruction For continuous use, store undiluted antibody at 2-8°C for up to a week. For long-term storage, aliquot and store at -20°C. -

Thrombocytopenia-Associated Mutations in Ser/Thr Kinase MASTL
Thrombocytopenia-associated mutations in Ser/Thr kinase MASTL deregulate actin cytoskeleton dynamics in platelets by Begoña Hurtado et al. SUPPLEMENTARY MATERIAL List of Supplementary Figures Figure S1. Hematopoietic precursors and megakaryocytes maturation in Mastl mutant mice. Figure S2. Sialylation and apoptosis profile of Mastl mutant platelets. Figure S3. Mastl E166D activity results in increased phosphorylation levels. Figure S4. Phospho-proteomic analysis in Mastl mutant platelets after thrombin activation. Figure S5. Signaling pathways differentially phosphorylated in Mastl mutant platelets. Figure S6. Changes in the phosphorylation status of signaling molecules in Mastl-mutant platelets. Figure S7. Full scan of blots showed in the manuscript. List of Supplementary Tables Table S1. Full data from phospho-proteomic studies. 1 Table S2. KEGG pathways enriched (FDR>0.05) in hyperphosphorylated proteins in resting Mastl(ED/ED) platelets (log2FC ED/WT>0.75), considering as statistical background the mouse platelet proteome. Table S3. KEGG pathways enriched (FDR>0.05) in hypophosphorylated proteins in resting Mastl(/) platelets (log2FC/WT<0.75), considering as statistical background the mouse platelet proteome. Table S4. KEGG pathways enriched (FDR>0.01) in hyperphosphorylated proteins in Mastl(ED/ED) platelets 3 minutes after stimulation with thrombin (log2FC ED/WT>0.75). Table S5. KEGG pathways enriched (FDR>0.01) in hyperphosphorylated proteins in both resting and 3-min-activated Mastl(ED/ED) platelets (log2FC ED/WT>0.5), considering as statistical background the mouse platelet proteome. Table S6. KEGG pathways enriched (FDR>0.05) in hyperphosphorylated proteins in Mastl(ED/ED) platelets 15 minutes after stimulation with thrombin (log2FC ED/WT>1.0), considering as statistical background the mouse platelet proteome. -

Pik3r1 Is Required for Glucocorticoid-Induced Perilipin 1 Phosphorylation in Lipid Droplet for Adipocyte Lipolysis
Diabetes Volume 66, June 2017 1601 Pik3r1 Is Required for Glucocorticoid-Induced Perilipin 1 Phosphorylation in Lipid Droplet for Adipocyte Lipolysis Taiyi Kuo,1,2 Tzu-Chieh Chen,2,3 Rebecca A. Lee,1,2 Nguyen Huynh Thao Nguyen,2 Augusta E. Broughton,2 Danyun Zhang,2 and Jen-Chywan Wang1,2,3 Diabetes 2017;66:1601–1610 | https://doi.org/10.2337/db16-0831 Glucocorticoids promote lipolysis in white adipose requiring tissues to produce ATP. However, prolonged GC tissue (WAT) to adapt to energy demands under stress, exposure results in dyslipidemia, hepatic steatosis, and whereas superfluous lipolysis causes metabolic dis- insulin resistance (1–3). Although exogenous GCs are fre- orders, including dyslipidemia and hepatic steatosis. quently prescribed as effective anti-inflammatory agents, Glucocorticoid-induced lipolysis requires the phosphor- their actions in metabolic tissues pose a therapeutic co- ylation of cytosolic hormone-sensitive lipase (HSL) and nundrum (4). perilipin 1 (Plin1) in the lipid droplet by protein kinase A GCs relay their message through the intracellular GC SIGNAL TRANSDUCTION fi p85a (PKA). We previously identi ed Pik3r1 (also called ) receptor (GR), which is a transcription factor. GC-induced as a glucocorticoid receptor target gene. Here, we adipocyte lipolysis requires de novo protein synthesis found that glucocorticoids increased HSL phosphoryla- (5–7), which is consistent with the concept that the GR tion, but not Plin1 phosphorylation, in adipose tissue- exerts its main function through modulating gene expres- specific Pik3r1-null (AKO) mice. Furthermore, in lipid sion. Several mechanisms are documented. First, the GCs droplets, the phosphorylation of HSL and Plin1 and the levels of catalytic and regulatory subunits of PKA were increase the transcription of genes encoding lipolytic increased by glucocorticoids in wild-type mice. -

The Role of Pik3r1 in the Regulation of Adipose Tissue Insulin
THE ROLE OF PIK3R1 IN THE REGULATION OF ADIPOSE TISSUE INSULIN SENSITIVITY by ZACHARY STEPHEN CLAYTON A DISSERTATION Presented to the Department of Human Physiology and the Graduate School of the University of Oregon in partial fulfillment of the requirements for the degree of Doctor of Philosophy June 2018 DISSERTATION APPROVAL PAGE Student: Zachary Stephen Clayton Title: The Role of Pik3r1 in the Regulation of Adipose Tissue Insulin Sensitivity This dissertation has been accepted and approved in partial fulfillment of the requirements for the Doctor of Philosophy degree in the Department of Human Physiology by: Carrie E. McCurdy, PhD Advisor John R. Halliwill, PhD Core Member Hans C. Dreyer, PhD Core Member Annie Zemper, PhD Institutional Representative and Sara D. Hodges Interim Vice Provost and Dean of the Graduate School Original approval signatures are on file with the University of Oregon Graduate School. Degree awarded June 2018. ii © 2018 Zachary Stephen Clayton iii DISSERTATION ABSTRACT Zachary Stephen Clayton Doctor of Philosophy Department of Human Physiology June 2018 Title: The Role of Pik3r1 in the Regulation of Adipose Tissue Insulin Sensitivity Obesity is a burgeoning health crisis in the United States. Obesity is associated with an earlier and greater risk for developing metabolic diseases. Insulin resistance is a central and defining feature of the metabolic diseases associated with obesity. Class 1a Phosphatidylinositol 3-kinase (PI3K) is integral in canonical insulin signaling. PI3K contains regulatory (p85a/b, p55a/g, p50a) and catalytic (p110a/b/d) subunits. The a regulatory subunits are encoded by Pik3r1. Increased Pik3r1 abundance has been observed in obese white adipose tissue (WAT). -

Pik3r1 Is Required for Glucocorticoid-Induced Perilipin 1 Phosphorylation
Page 1 of 44 Diabetes Pik3r1 is Required for Glucocorticoid-induced Perilipin 1 Phosphorylation in Lipid Droplet for Adipocyte Lipolysis Taiyi Kuo1, 3+, Tzu-Chieh Chen2,3+, Rebecca A. Lee1,3, Nguyen Huynh Thao Nguyen3, Augusta E 3 3 1, 2, 3* Broughton , Danyun Zhang , and Jen-Chywan Wang Endocrinology Graduate Program1, Metabolic Biology Graduate Program2 and Department of Nutritional Sciences & Toxicology3, University of California Berkeley, Berkeley, CA 94720- 3104 + These authors contributed equally to this work *Corresponding author: Jen-Chywan Wang, 315 Morgan Hall, UC Berkeley, Berkeley, CA 94720-3104, email: [email protected], Phone: 510-643-1039 Running Title: Pik3r1 and glucocorticoid-induced adipocyte lipolysis Keywords: Glucocorticoids, Glucocorticoid Receptor, Lipolysis, Pik3r1, Perilipin 1, Protein Kinase A 1 Diabetes Publish Ahead of Print, published online March 14, 2017 Diabetes Page 2 of 44 Abstract Glucocorticoids promote lipolysis in white adipose tissue (WAT) to adapt to energy demands under stress, while superfluous lipolysis causes metabolic disorders, including dyslipidemia and hepatic steatosis. Glucocorticoid-induced lipolysis requires the phosphorylation of cytosolic hormone sensitive lipase (HSL) and perilipin 1 (Plin1) in the lipid droplet by protein kinase A (PKA). We previously identified Pik3r1 (a.k.a. p85α) as a glucocorticoid receptor target gene. Here, we found that glucocorticoids increased HSL phosphorylation, but not Plin1 phosphorylation, in adipose tissue-specific Pik3r1-null (AKO) mice. Furthermore, in lipid droplets, the phosphorylation of HSL and Plin1 and the levels of catalytic and regulatory subunits of PKA were increased by glucocorticoids in wild type mice. However, these effects were attenuated in AKO mice. In agreement with reduced WAT lipolysis, glucocorticoid- initiated hepatic steatosis and hypertriglyceridemia were improved in AKO mice. -

Integrative Kinome Profiling Identifies Mtorc1/2 Inhibition As Treatment Strategy in Ovarian Clear Cell Carcinoma
Published OnlineFirst April 23, 2018; DOI: 10.1158/1078-0432.CCR-17-3060 Cancer Therapy: Preclinical Clinical Cancer Research Integrative Kinome Profiling Identifies mTORC1/2 Inhibition as Treatment Strategy in Ovarian Clear Cell Carcinoma Joseph J. Caumanns1, Katrien Berns2, G. Bea A. Wisman1, Rudolf S.N. Fehrmann3, Tushar Tomar1, Harry Klip1, Gert J. Meersma1, E. Marielle Hijmans2, Annemiek M.C. Gennissen2, Evelien W. Duiker4, Desiree Weening5, Hiroaki Itamochi6, Roelof J.C. Kluin2, Anna K.L. Reyners3, Michael J. Birrer7, Helga B. Salvesen8, Ignace Vergote9, Els van Nieuwenhuysen9, James Brenton10, E. Ioana Braicu11, Jolanta Kupryjanczyk12, Beata Spiewankiewicz13, Lorenza Mittempergher2, Rene Bernards2, Ate G.J. van der Zee1, and Steven de Jong3 Abstract Purpose: Advanced-stage ovarian clear cell carcinoma Results: We identified several putative driver mutations in (OCCC) is unresponsive to conventional platinum-based che- kinases at low frequency that were not previously annotated in motherapy. Frequent alterations in OCCC include deleterious OCCC. Combining mutations and copy-number alterations, 91% mutations in the tumor suppressor ARID1A and activating of all tumors are affected in the PI3K/AKT/mTOR pathway, mutations in the PI3K subunit PIK3CA.Inthisstudy,weaimed the MAPK pathway, or the ERBB family of receptor tyrosine to identify currently unknown mutated kinases in patients with kinases, and 82% in the DNA repair pathway. Strong p-S6 staining OCCC and test druggability of downstream affected pathways in patients with OCCC suggests high mTORC1/2 activity. We in OCCC models. consistently found that the majority of OCCC cell lines are Experimental Design: In a large set of patients with OCCC (n ¼ especially sensitive to mTORC1/2 inhibition by AZD8055 and 124), the human kinome (518 kinases) and additional cancer- not toward drugs targeting ERBB family of receptor tyrosine related genes were sequenced, and copy-number alterations were kinases or DNA repair signaling. -

Attenuated Pik3r1 Expression Prevents Insulin Resistance and Adipose Tissue Macrophage Accumulation in Diet-Induced Obese Mice Carrie E
ORIGINAL ARTICLE Attenuated Pik3r1 Expression Prevents Insulin Resistance and Adipose Tissue Macrophage Accumulation in Diet-Induced Obese Mice Carrie E. McCurdy,1,2,3 Simon Schenk,4 Michael J. Holliday,1,2,3 Andrew Philp,5 Julie A. Houck,1,2,3 David Patsouris,6 Paul S. MacLean,2 Susan M. Majka,2,3 Dwight J. Klemm,2,3 and Jacob E. Friedman1,7 Obese white adipose tissue (AT) is characterized by large-scale inhibitor of kappa B kinase b (IKKb)andJunNH2-terminal infiltration of proinflammatory macrophages, in parallel with sys- kinase (JNK), which impinge upon the insulin signaling temic insulin resistance; however, the cellular stimulus that cascade by inhibiting tyrosine phosphorylation of insulin initiates this signaling cascade and chemokine release is still receptor substrate 1 (IRS1), leading to impaired insulin unknown. The objective of this study was to determine the role of activation of phosphoinositide 3-kinase (PI3K) and Akt (1). the phosphoinositide 3-kinase (PI3K) regulatory subunits on AT The chemoattractant stimulus and the molecular details fi fi macrophage (ATM) in ltration in obesity. Here, we nd that the underlying cross-talk between infiltrating macrophage and Pik3r1 regulatory subunits (i.e., p85a/p55a/p50a) are highly in- duced in AT from high-fat diet–fed obese mice, concurrent with the adipocyte is an area of intense investigation. Envi- insulin resistance. Global heterozygous deletion of the Pik3r1 reg- ronmental cues including adipose tissue (AT) hypoxia, cell ulatory subunits (aHZ), but not knockout of Pik3r2 (p85b), pre- death, physical stress on adipocyte extracellular matrix, serves whole-body, AT, and skeletal muscle insulin sensitivity, and increased lipolysis have all been identified as mecha- despite severe obesity. -

Kinome Profiling to Predict Sensitivity to MAPK Inhibition in Melanoma
cancers Article Kinome Profiling to Predict Sensitivity to MAPK Inhibition in Melanoma and to Provide New Insights into Intrinsic and Acquired Mechanism of Resistance 1, , 2, 1 3 Mohamad Krayem * y , Philippe Aftimos y, Ahmad Najem , Tim van den Hooven , Adriënne van den Berg 3, Liesbeth Hovestad-Bijl 3, Rik de Wijn 3, Riet Hilhorst 3 , Rob Ruijtenbeek 3, Malak Sabbah 1, Joseph Kerger 2, Ahmad Awada 1,2 , Fabrice Journe 1 and Ghanem E. Ghanem 1 1 Laboratory of Oncology and Experimental Surgery, Institut Jules Bordet, Université Libre de Bruxelles, 1000 Brussels, Belgium; [email protected] (A.N.); [email protected] (M.S.); [email protected] (A.A.); [email protected] (F.J.); [email protected] (G.E.G.) 2 Medical Oncology Clinic, Institut Jules Bordet, Université Libre de Bruxelles, 1000 Brussels, Belgium; [email protected] (P.A.); [email protected] (J.K.) 3 PamGene International BV, 5211HH ’s-Hertogenbosch, The Netherlands; [email protected] (T.v.d.H.); [email protected] (A.v.d.B.); [email protected] (L.H.-B.); [email protected] (R.d.W.); [email protected] (R.H.); [email protected] (R.R.) * Correspondence: [email protected] Both authors equally contributed to this manuscript. y Received: 20 January 2020; Accepted: 20 February 2020; Published: 22 February 2020 Abstract: Mitogen-activated protein kinase (MAPK) inhibition with the combination of BRAF (Rapidly Accelerated Fibrosarcoma) and MEK (Mitogen-activated protein kinase kinase) inhibitors has become the standard of first-line therapy of metastatic melanoma harbouring BRAF V600 mutations. However, about half of the patients present with primary resistance while the remaining develop secondary resistance under prolonged treatment. -
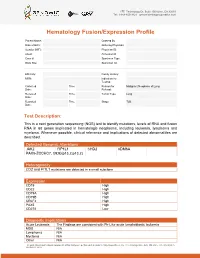
Sample Report for Hematology Fusion/Expression Profile
17527 Technology Dr, Suite 100 Irvine, CA 92618 Tel: 1-949-450-9421 genomictestingcooperative.com Hematology Fusion/Expression Profile Patient Name: Ordered By Date of Birth: Ordering Physician: Gender (M/F): Physician ID: Client: Accession #: Case #: Specimen Type: Body Site: Specimen ID: _____________________________________________________________________________________________ Ethnicity: Family History: MRN: Indication for Testing: Collected Time Reason for Malignant Neoplasm of Lung Date: : Referral: Received Time Tumor Type: Lung Date: : Reported Time Stage: T2B Date: : Test Description: This is a next generation sequencing (NGS) test to identify mutations, levels of RNA and fusion RNA in 68 genes implicated in hematologic neoplasms, including leukemia, lymphoma and myeloma. Whenever possible, clinical relevance and implications of detected abnormalities are described. Detected Genomic Alterations JAK2 FIP1L1 CHD2 KDM6A PAX5-ZCCHC7, t(9;9)(p13.2;p13.2) Heterogeneity CD2 and FI1L1 mutations are detected in a small subclone Expression CD19 High CD22 High CD79A High CD79B High CRLF2 High PAX5 High CD274 Low Diagnostic Implications Acute Leukemia The Findings are consistent with Ph-Like acute lymphoblastic leukemia MDS N/A Lymphoma N/A Myeloma N/A Other N/A The professional and technical components of this assay were performed at Genomic Testing Cooperative, LCA, 27 Technology Drive, Suite 100, Irvine, CA 92618 (CLIA ID: 05D2111917). The assay is FDA cleared and the performance characteristics were established at this location and was .... 27 Technology Dr, Suite 100 Irvine, CA 92618 Tel: 1-949-450-9421 genomictestingcooperative.com Therapeutic Implications JAK2 Momelotinib and ruxolitinib Prognostic Implications JAK2 Poor CRLF2 Poor Overall Poor Relevant Genes with No Alteration None Results Summary • Fusion (PAX5-ZCCHC7) transcript involving PAX5 and ZCCHC7 genes resulting from t(9;9)(p13.2;p13.2) translocation is detected.