Synthesis, Characterization and Thermal Decomposition Of
Total Page:16
File Type:pdf, Size:1020Kb

Load more
Recommended publications
-

Problem Formulation of the Risk Evaluation for Perchloroethylene (Ethene, 1,1,2,2-Tetrachloro)
EPA Document# EPA-740-R1-7017 May 2018 DRAFTUnited States Office of Chemical Safety and Environmental Protection Agency Pollution Prevention Problem Formulation of the Risk Evaluation for Perchloroethylene (Ethene, 1,1,2,2-Tetrachloro) CASRN: 127-18-4 May 2018 TABLE OF CONTENTS ABBREVIATIONS ............................................................................................................................ 8 EXECUTIVE SUMMARY .............................................................................................................. 11 1 INTRODUCTION .................................................................................................................... 14 1.1 Regulatory History ..................................................................................................................... 16 1.2 Assessment History .................................................................................................................... 16 1.3 Data and Information Collection ................................................................................................ 18 1.4 Data Screening During Problem Formulation ............................................................................ 19 2 PROBLEM FORMULATION ................................................................................................. 20 2.1 Physical and Chemical Properties .............................................................................................. 20 2.2 Conditions of Use ...................................................................................................................... -

Fluorinated Polymers As Smart Materials for Advanced Biomedical Applications
polymers Review Fluorinated Polymers as Smart Materials for Advanced Biomedical Applications Vanessa F. Cardoso 1,2,* ID , Daniela M. Correia 3,4, Clarisse Ribeiro 1,5 ID , Margarida M. Fernandes 1,5 and Senentxu Lanceros-Méndez 4,6 1 Centro/Departamento de Física, Universidade do Minho, 4710-057 Braga, Portugal; cribeiro@fisica.uminho.pt (C.R.); margaridafernandes@fisica.uminho.pt (M.M.F.) 2 CMEMS-UMinho, Universidade do Minho, DEI, 4800-058 Guimaraes, Portugal 3 Departamento de Química e CQ-VR, Universidade de Trás-os-Montes e Alto Douro, 5001-801 Vila Real, Portugal; [email protected] 4 BCMaterials, Basque Center for Materials, Applications and Nanostructures, UPV/EHU Science Park, 48940 Leioa, Spain; [email protected] 5 CEB—Centre of Biological Engineering, University of Minho, 4710-057 Braga, Portugal 6 IKERBASQUE, Basque Foundation for Science, 48013 Bilbao, Spain * Correspondence: [email protected]; Tel.: +351-253-60-40-73 Received: 11 January 2018; Accepted: 6 February 2018; Published: 8 February 2018 Abstract: Fluorinated polymers constitute a unique class of materials that exhibit a combination of suitable properties for a wide range of applications, which mainly arise from their outstanding chemical resistance, thermal stability, low friction coefficients and electrical properties. Furthermore, those presenting stimuli-responsive properties have found widespread industrial and commercial applications, based on their ability to change in a controlled fashion one or more of their physicochemical properties, in response to single or multiple external stimuli such as light, temperature, electrical and magnetic fields, pH and/or biological signals. In particular, some fluorinated polymers have been intensively investigated and applied due to their piezoelectric, pyroelectric and ferroelectric properties in biomedical applications including controlled drug delivery systems, tissue engineering, microfluidic and artificial muscle actuators, among others. -

Chlorotrifluoroethylene Interim AEGL Document
ACUTE EXPOSURE GUIDELINE LEVELS (AEGLs) FOR CHLOROTRIFLUOROETHYLENE (CAS Reg. No. 79-38-9) CF2 = CFCl INTERIM ACUTE EXPOSURE GUIDELINE LEVELS (AEGLs) FOR CHLOROTRIFLUOROETHYLENE (CAS Reg. No. 79-38-9) INTERIM CHLOROTRIFLUOROETHYLENE INTERIM: 06/2008/ Page 3 of 34 PREFACE Under the authority of the Federal Advisory Committee Act (FACA) P. L. 92-463 of 1972, the National Advisory Committee for Acute Exposure Guideline Levels for Hazardous Substances (NAC/AEGL Committee) has been established to identify, review and interpret relevant toxicologic and other scientific data and develop AEGLs for high priority, acutely toxic chemicals. AEGLs represent threshold exposure limits for the general public and are applicable to emergency exposure periods ranging from 10 minutes to 8 hours. Three levels – AEGL-1, AEGL-2 and AEGL-3 C are developed for each of five exposure periods (10 and 30 minutes, 1 hour, 4 hours, and 8 hours) and are distinguished by varying degrees of severity of toxic effects. The three AEGLs are defined as follows: AEGL-1 is the airborne concentration (expressed as parts per million or milligrams per cubic meter [ppm or mg/m3]) of a substance above which it is predicted that the general population, including susceptible individuals, could experience notable discomfort, irritation, or certain asymptomatic, non-sensory effects. However, the effects are not disabling and are transient and reversible upon cessation of exposure. AEGL-2 is the airborne concentration (expressed as ppm or mg/m3) of a substance above which it is predicted that the general population, including susceptible individuals, could experience irreversible or other serious, long-lasting adverse health effects or an impaired ability to escape. -

20210311 IAEG AD-DSL V5.0 for Pdf.Xlsx
IAEGTM AD-DSL Release Version 4.1 12-30-2020 Authority: IAEG Identity: AD-DSL Version number: 4.1 Issue Date: 2020-12-30 Key Yellow shading indicates AD-DSL family group entries, which can be expanded to display a non-exhaustive list of secondary CAS numbers belonging to the family group Substance Identification Change Log IAEG Regulatory Date First Parent Group IAEG ID CAS EC Name Synonyms Revision Date ECHA ID Entry Type Criteria Added IAEG ID IAEG000001 1327-53-3 215-481-4 Diarsenic trioxide Arsenic trioxide R1;R2;D1 2015-03-17 2015-03-17 100.014.075 Substance Direct Entry IAEG000002 1303-28-2 215-116-9 Diarsenic pentaoxide Arsenic pentoxide; Arsenic oxide R1;R2;D1 2015-03-17 2015-03-17 100.013.743 Substance Direct Entry IAEG000003 15606-95-8 427-700-2 Triethyl arsenate R1;R2;D1 2015-03-17 2017-08-14 100.102.611 Substance Direct Entry IAEG000004 7778-39-4 231-901-9 Arsenic acid R1;R2;D1 2015-03-17 2015-03-17 100.029.001 Substance Direct Entry IAEG000005 3687-31-8 222-979-5 Trilead diarsenate R1;R2;D1 2015-03-17 2017-08-14 100.020.890 Substance Direct Entry IAEG000006 7778-44-1 231-904-5 Calcium arsenate R1;R2;D1 2015-03-17 2017-08-14 100.029.003 Substance Direct Entry IAEG000009 12006-15-4 234-484-1 Cadmium arsenide Tricadmium diarsenide R1;R2;D1 2017-08-14 2017-08-14 Substance Direct Entry IAEG000021 7440-41-7 231-150-7 Beryllium (Be) R2 2015-03-17 2019-01-24 Substance Direct Entry IAEG000022 1306-19-0 215-146-2 Cadmium oxide R1;R2;D1 2015-03-17 2017-08-14 100.013.770 Substance Direct Entry IAEG000023 10108-64-2 233-296-7 Cadmium -

1,1,2-Trichloro-1,2,2-Trifluoroethane
1,1,2-Trichloro-1,2,2-trifluoroethane sc-251541 Material Safety Data Sheet Hazard Alert Code Key: EXTREME HIGH MODERATE LOW Section 1 - CHEMICAL PRODUCT AND COMPANY IDENTIFICATION PRODUCT NAME 1,1,2-Trichloro-1,2,2-trifluoroethane STATEMENT OF HAZARDOUS NATURE CONSIDERED A HAZARDOUS SUBSTANCE ACCORDING TO OSHA 29 CFR 1910.1200. NFPA FLAMMABILITY0 HEALTH3 HAZARD INSTABILITY1 SUPPLIER Company: Santa Cruz Biotechnology, Inc. Address: 2145 Delaware Ave Santa Cruz, CA 95060 Telephone: 800.457.3801 or 831.457.3800 Emergency Tel: CHEMWATCH: From within the US and Canada: 877-715-9305 Emergency Tel: From outside the US and Canada: +800 2436 2255 (1-800-CHEMCALL) or call +613 9573 3112 PRODUCT USE Used for cleaning, degreasing electrical equipment; a dry-cleaning solvent; in manufacture of chlorotrifluoroethylene; in liquid fire extinguishers, as a urethane foam blowing agent, refrigerant. Ozone depletion potential 1.0/1.0 Severe. The Montreal Protocol on Substances that Deplete the Ozone Layer has caused a substantial decline in its use. SYNONYMS C2-Cl3-F3, ClF2C-CCl2F, CFC-113, CFC113, trichlorotrifluoroethane, "1, 1, 2-trichlorotrifluoroethane", "1, 2, 2-trichlorotrifluoroethane", "1, 1, 2-trichloro-1, 2, 2-trifluoroethane", "1, 1, 2-trifluorotrichloroethane", "1, 1, 2-trifluoro-1, 2, 2-trichloroethane", Arcton-63, Arklone-P, Asahifron-113, Daiflon-S3, F-113, FC-113, Fluorocarbon-113, Forane, Freon-113, Frigen-113A, Frigen-113-TR, Frigen-113-TR-N, Frigen- 113-TR-T, Genetron-113, Halocarbon-113, Isceon-113, "Kaiser Chemicals-11", Khladon-113, Ledon-113, R-113, "Refrigerant R-113", "R-113 (Halocarbon)", Ucon-113, "Ucon Fluorocarbon-113", "Ucon 113/Halocarbon-113" Section 2 - HAZARDS IDENTIFICATION CHEMWATCH HAZARD RATINGS Min Max Flammability: 0 Toxicity: 2 Min/Nil=0 Body Contact: 4 Low=1 Moderate=2 Reactivity: 1 High=3 Extreme=4 1 of 16 Chronic: 2 CANADIAN WHMIS SYMBOLS EMERGENCY OVERVIEW RISK Forms very sensitive explosive metallic compounds. -
Table for Chemistry Compatibility
ETFE TabLE foR Head cover and clamping chemistRY compatibiLitY disc are made of thick PTFE ETFE/ FFKM walled molded ETFE with ECTFE metal core for unsurpassed Acid amides ++ ++ ++ Dimethylformamide (DMF), Acetamide, Formamide long-term performance . Acids, dilute or weak ++ ++ ++ . Acetic acid, Carbonic acid, Butyric acid DATA TERATURE NG I Acids, strong or concentrated ++ ++ ++ E S PTFE Hydrochloric acid, Sulfuric acid, Nitric acid, S LI THE UENC OU Trifluoroacetic acid (TFA) I FL ON N Alcohols, aliphatic VAR ++ ++ ++ I E Methanol, Ethanol, Butanol L B Well-proven PTFE ANCE ROM LI SSI Aldehydes ++ ++ ++ F O sandwich diaphragms for RE Formaldehyde, Ethanal, Hexanal ON I F P increased reliability and ER O S U Amines ++ ++ + Y extended service life N-Methyl-2-pyrrolidone (NMP), Triethylamine ET I ORMAT F ROM N VAR F I Bases ++ ++ ++ Sodium hydroxide, Potassium hydroxide, Ammonia ON VED THE I ED TO Esters S ++ ++ ++ FFKM DER Ethyl acetate, Butyl formate, Amyl butyrate BA UE BE Y . D ARE Ethers ++ ++ ++ MA Diethyl ether, Tetrahydrofurane, Dioxane S DATA Inlet and outlet flapper M DATA E I S Hydrocarbons, aliphatic A ++ ++ ++ L valves are made of FFKM HE C Pentane, Hexane, Heptane THE L or PTFE for both highest F O Hydrocarbons, aromatic ++ ++ ++ EGA leakage tightness and best Y Benzene, Toluene, Xylene L chemical resistance O Hydrocarbons, halogenated ++ ++ ++ . N DE I Methyl chloride, Chloroform, Ethylene chloride ACCURAC GU A Ketones THE ++ ++* ++ S ECTFE Acetone, Cyclohexanone A Oxidizing acids, oxidizing agents ++ + ++ LY ARRANT ON Ozone, Hydrogen peroxide, Chlorine W ERVE Sulfoxides ++ ++ ++ NOT Valve connectors are made S CHEMISTRY COMPATIBILITY of ECTFE for an optimum Dimethyl sulfoxide (DMSO) S CAN of VACUUBRAND Chemistry Diaphragm Pumps DOE of thermal, mechanical and Y chemical resistance THE POLYTETRAFLUOROETHYLENE exceLLENT CHEMICAL RESISTANCE PTFE: ++ S, ETFE: ETHYLENE TETRAFLUOROETHYLENE + good TO LIMITED CHEMICAL ECTFE: ETHYLENE RESISTANCE CHLOROTRIFLUOROETHYLENE ACTOR ©VACUUBRAND GMBH + CO KG . -
C1-C4 Halogenated HC Info 16FEB2018.Xlsx
C1-C4 Halogenated Hydrocarbons/Halocarbons Not Otherwise Listed (C1-C4 NOL) Feb 28, 2018 Administrative Council Mtg - Printed 2/16/20184:11 PM Reported Uses (SOURCES:HAZMAP, NJDHSS, Wikipedia, TSCA CDR, mfr literature) TURA TSCA CDR Chemical CAS/Chemica chemical name chemical fire 2015 Chemical Name Pesti- blowing feedstock / supressant / propell- Inventory? TRI? List l Number (synonyms) formula Solvent refriger-ant etchant Other Tier II cide agent intermed-iate flame ant status* retardant insulation for refrigerators, freezers, commercial refrigeration equipment, refrigerated containers and LNG ships; spray foam insulation; insulated metal panels; slabstock and molded flexible foam; refrigerant for chillers; and solvents for metal cleaning and electronics, and circuit flush. https://www.honeywell-blowingagents.com/?document=solstice-lba-technical- HFO-1233zd (E) brochure&download=1 Hydro-fluoro-olefin C H ClF YYY yhttps://www.honeywell-refrigerants.com/americas/product/solstice-zd/ (HFO) 3 2 3 CDR 2016 Honeywell: 100% as propellants and blowing agents for plastics product mfg; R-1233zd CDR Honeywell Solstice® ZD refrigerant, Solstice® 1233zdE, Solstice Blowing Agent; Arkema (current Forane® 1233zd blowing agent for spray PU foam, appliance insulation, etc. and solvent (may Honeywell not be available in US) product) C1-C4 Cat 102687-65-0 1-Chloro-3,3,3-trifluoropropene Y 106‐95‐6 Allyl bromide 2-Propenyl bromide C3H5Br Y y Manufacture of synthetic perfumes, other allyl compounds; Insecticidal fumigant; 1-Propene, 3-bromo- Chemical intermediate in organic synthesis, for resins (copolymer with sulfur dioxide) and fragrances; As a fumigant (if quite volatile) or as a contact poison; C1-C4 Cat Used in the manufacture of plastics and dyestuff. -
Properties and Hazards of 108 Selected Substances -1992 Edition
U. S. DEPARTMENT OF THE INTERIOR GEOLOGICAL SURVEY PROPERTIES AND HAZARDS OF 108 SELECTED SUBSTANCES -1992 EDITION Jeffrey E. Lucius1, Gary R. Olhoeft1, Patricia L. Hill1, and Steven K. Duke2 U.S. Geological Survey Open-File Report 92-527 September 1992 This report is preliminary and has not been reviewed for conformity with U.S. Geological Survey editorial standards. Any use of trade, product, or firm names is for descriptive purposes only and does not imply endorsement by the U.S. Government. 1 Golden, CO, USA 2 now with Woodward-Clyde Consultants, 2020 E. First Street, Suite 900, Santa Ana, CA 92705 iii CONTENTS List of Tables ....................................................... vi Notice ............................................................... vii Introduction ......................................................... 1 Properties Definitions ................................................... 4 Key to Abbreviations .......................................... 13 Conversion Factors ............................................ 16 Tables .............................................................. 18 Substances .............................. GAS RN Acetic acid .................... 64-19-7 .................. 70 Acetone ........................ 67-64-1 .................. 75 Acrolein ....................... 107-02-8 .................. 81 Acrylonitrile .................. 107-13-1 .................. 85 Aldrin ......................... 309-00-2 .................. 89 Ammonia ........................ 7664-41-7 .................. 92 -
Strategy for Chemical Analysis of Alternative Refrigerants
UNITED STATES DEPARTMENT OF COMMERCE NATIONAL INSTITUTE OF STANDARDS AND TECHNOLOGY NisrNATL INST. OF STAND & TECH R.I.C. A111D3 MblfiD? NIST PUBLICATIONS NIST Technical Note 1340 strategy for Chemical Analysis l^^ "^ of Alternative Refrigerants ^ Thomas J. Bruno Thermophysics Division Center for Cinemical Technology National Measurement Laboratory National Institute of Standards and Technology Boulder, Colorado 80303-3328 \. U.S. DEPARTMENT OF COMMERCE, Robert A. Mosbacher, Secretary NATIONAL INSTITUTE OF STANDARDS AND TECHNOLOGY, John W. Lyons, Director Issued August 1990 National Institute of Standards and Technology Technical Note 1340 Natl. Inst. Stand. Technol., Tech. Note 1340, 104 pages (Aug. 1990) CODEN:NTNOEF U.S. GOVERNMENT PRINTING OFFICE WASHINGTON: 1990 For sale by the Superintendent of Documents, U.S. Government Printing Office, Washington, DC 20402-9325 CONTENTS Page Abstract 1 1. INTRODUCTION 1 2. QUALITATIVE IDENTIFICATION 7 2.1 Mass Spectra 9 2.2 Infrared Spectophotometry 20 2.3 Adsorbent Tests 22 3. QUANTITATION AND SEPARATION METHODS 27 3.1 Analysis of Organics 27 3.2 Analysis of Air 31 3.3 Analysis of Water 40 4. REACTION SCREENING 44 5. ACKNOWLEDGMENTS 48 6. REFERENCES 48 Appendix 1. Sources of toxicology and physical properties 51 Appendix 2. Computer program to aid in the interpretation of mass spectra especially in the analysis of chlorofluoroethanes and -ethylenes 53 Appendix 3. Vortex-cooled chromatographic syringe 87 Appendix 4. Refrigerant code numbers for ethane- and ethylene-based compounds 95 m List of Tables Page Table 1. List of compounds studied in the development of spectral library and in the study of spectral trends 7 Table 2. -

Chlorotrifluoroethylene Copolymer Chlortrifluorethylen-Copolymer Copolymère Polymonochlorotrifluoré
(19) TZZ_74_¥ _T (11) EP 1 741 732 B1 (12) EUROPEAN PATENT SPECIFICATION (45) Date of publication and mention (51) Int Cl.: of the grant of the patent: C08F 214/24 (2006.01) 19.06.2013 Bulletin 2013/25 (86) International application number: (21) Application number: 05730418.0 PCT/JP2005/007133 (22) Date of filing: 13.04.2005 (87) International publication number: WO 2005/100420 (27.10.2005 Gazette 2005/43) (54) CHLOROTRIFLUOROETHYLENE COPOLYMER CHLORTRIFLUORETHYLEN-COPOLYMER COPOLYMÈRE POLYMONOCHLOROTRIFLUORÉ (84) Designated Contracting States: • KITAHARA, Takahiro AT BE BG CH CY CZ DE DK EE ES FI FR GB GR DAIKIN INDUSTRIES, LTD. HU IE IS IT LI LT LU MC NL PL PT RO SE SI SK TR Settsu-shi, Osaka 5668585 (JP) (30) Priority: 13.04.2004 JP 2004118365 (74) Representative: HOFFMANN EITLE Patent- und Rechtsanwälte (43) Date of publication of application: Arabellastrasse 4 10.01.2007 Bulletin 2007/02 81925 München (DE) (73) Proprietor: DAIKIN INDUSTRIES, LTD. (56) References cited: Osaka-shi, Osaka 530-8323 (JP) WO-A1-2004/098880 JP-A- 54 132 691 JP-A- 63 307 609 JP-A- 2002 503 744 (72) Inventors: JP-A- 2004 277 689 JP-B1- 48 025 415 • SHIMONO, Takeshi US-A- 3 331 811 US-B1- 6 177 196 DAIKIN INDUSTRIES, LTD. Settsu-shi, Osaka 5668585 (JP) • DATABASE CAPLUS [Online] HABERMANN, • AOYAMA, Takahisa KENNET R: ’Laminated polymer sheets’, DAIKIN INDUSTRIES, LTD. XP008125868 Retrieved from STN Database Settsu-shi, Osaka 5668585 (JP) accession no. 1967:38567 & BE 670 586 A 31 • TORIMAE, Hiroshi January 1966 DAIKIN INDUSTRIES, LTD. Settsu-shi, Osaka 5668585 (JP) Note: Within nine months of the publication of the mention of the grant of the European patent in the European Patent Bulletin, any person may give notice to the European Patent Office of opposition to that patent, in accordance with the Implementing Regulations. -
Brooks® Model 585Xemh High Temperature Mass Flow Controllers
Installation and Operation Manual X-TMF-585xEMH-MFC-eng Part Number: 541B212AAG May, 2017 Brooks® Model 585xEMH Brooks® Model 585xEMH High Temperature Mass Flow Controllers Model 5850EMH High Temperature Model 5851EMH High Temperature Thermal Mass Flow Controller Thermal Mass Flow Controller Installation and Operation Manual X-TMF-585xEMH-MFC-eng Part Number: 541B212AAG Brooks® Model 585xEMH May, 2017 Essential Instructions Read before proceeding! Brooks Instrument designs, manufactures and tests its products to meet many national and international standards. These products must be properly installed, operated and maintained to ensure they continue to operate within their normal specifications. The following instructions must be adhered to and integrated into your safety program when installing, operating and maintaining Brooks Instrument products. • To ensure proper performance, use qualified personnel to install, operate, update, program and maintain the product. • Read all instructions prior to installing, operating and servicing the product. If this instruction manual is not the correct manual, please see back cover for local sales office contact information. Save this instruction manual for future reference. WARNING: Do not operate this instrument in excess of the specifications listed in the Instruction and Operation Manual. Failure to heed this warning can result in serious personal injury and / or damage to the equipment. • If you do not understand any of the instructions, contact your Brooks Instrument representative for clarification. • Follow all warnings, cautions and instructions marked on and supplied with the product. WARNING: Prior to installation ensure this instrument has the required approval ratings to meet local and national codes. Failure to heed this warning can result in serious personal injury and / or damage to the equipment. -
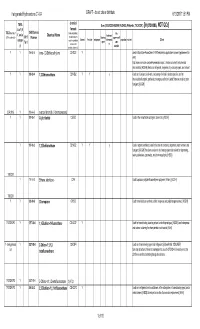
List for Comments C1-C4 Halogenated Hydrocarbons
Halogenated Hydrocarbons C1-C4 DRAFT - do not cite or distribute 6/13/201711:51 PM chemical TURA Uses (SOURCES:HAZMAP, NJDHSS, Wikipedia, TSCA CDR ) [In process, NOT QCd] List? (If formula TSCA Inventory? CAS/Chemica (Yellow highlighting = fire Y, not in Chemical Name feedstock/ (Y/N - student ck) 2015 l Number multiple halogens blowing supressant/fl Solvent Pesticide refrigerant intermedia propellant etchant Other categor could be substituted agent ame Tier II to create other te y) retardant possible substances) Y Y 156-60-5 trans-1,2-Dichloroethylene C2H2Cl2 Y used in MicroCare PowerClean II PW2 electronics applications solvent (replacement for nPB) http://www.microcare.com/p-64-powerclean.aspx; Used as a solvent and chemical intermediate; [ACGIH] Used as a refrigerant, degreaser, dry cleaning agent, and solvent YY 106-93-4 1,2-Dibromoethane C2H4Br2 Y Y y Usedfor perfumes, as a fumigant, adhesives, a solvent, lacquers, a scavenger oils, and forresins; lead [HSDB]in leaded gasoline, and an intermediate in organic synthesis; no longer used in the United States as a soil or grain fumigant; [ACGIH] CDR 2015 Y 106-94-5 n-propyl bromide (1-bromopropane) YY 107-05-1 Allyl chloride C3H5Cl Used in the manufacture of organic chemicals; [ACGIH] Y 107-06-2 1,2-Dichloroethane C2H4Cl2 Y Y y Used in organic synthesis; used in the past as a solvent, degreaser, paint remover, and fumigant; [ACGIH] Has been used as a dry cleaning agent and solvent for degreasing, resins, adhesives, coasmetics, and pharmaceuticals; [HSDB] TRI/CDR Y 116-14-3 Ethene, tetrafluoro-