Coleoptera: Cerambycidae) and Phylogenetic Relationships Within Cerambycidae
Total Page:16
File Type:pdf, Size:1020Kb

Load more
Recommended publications
-

Green-Tree Retention and Controlled Burning in Restoration and Conservation of Beetle Diversity in Boreal Forests
Dissertationes Forestales 21 Green-tree retention and controlled burning in restoration and conservation of beetle diversity in boreal forests Esko Hyvärinen Faculty of Forestry University of Joensuu Academic dissertation To be presented, with the permission of the Faculty of Forestry of the University of Joensuu, for public criticism in auditorium C2 of the University of Joensuu, Yliopistonkatu 4, Joensuu, on 9th June 2006, at 12 o’clock noon. 2 Title: Green-tree retention and controlled burning in restoration and conservation of beetle diversity in boreal forests Author: Esko Hyvärinen Dissertationes Forestales 21 Supervisors: Prof. Jari Kouki, Faculty of Forestry, University of Joensuu, Finland Docent Petri Martikainen, Faculty of Forestry, University of Joensuu, Finland Pre-examiners: Docent Jyrki Muona, Finnish Museum of Natural History, Zoological Museum, University of Helsinki, Helsinki, Finland Docent Tomas Roslin, Department of Biological and Environmental Sciences, Division of Population Biology, University of Helsinki, Helsinki, Finland Opponent: Prof. Bengt Gunnar Jonsson, Department of Natural Sciences, Mid Sweden University, Sundsvall, Sweden ISSN 1795-7389 ISBN-13: 978-951-651-130-9 (PDF) ISBN-10: 951-651-130-9 (PDF) Paper copy printed: Joensuun yliopistopaino, 2006 Publishers: The Finnish Society of Forest Science Finnish Forest Research Institute Faculty of Agriculture and Forestry of the University of Helsinki Faculty of Forestry of the University of Joensuu Editorial Office: The Finnish Society of Forest Science Unioninkatu 40A, 00170 Helsinki, Finland http://www.metla.fi/dissertationes 3 Hyvärinen, Esko 2006. Green-tree retention and controlled burning in restoration and conservation of beetle diversity in boreal forests. University of Joensuu, Faculty of Forestry. ABSTRACT The main aim of this thesis was to demonstrate the effects of green-tree retention and controlled burning on beetles (Coleoptera) in order to provide information applicable to the restoration and conservation of beetle species diversity in boreal forests. -

Data on Cerambycidae and Chrysomelidae (Coleoptera: Chrysomeloidea) from Bucureªti and Surroundings
Travaux du Muséum National d’Histoire Naturelle © Novembre Vol. LI pp. 387–416 «Grigore Antipa» 2008 DATA ON CERAMBYCIDAE AND CHRYSOMELIDAE (COLEOPTERA: CHRYSOMELOIDEA) FROM BUCUREªTI AND SURROUNDINGS RODICA SERAFIM, SANDA MAICAN Abstract. The paper presents a synthesis of the data refering to the presence of cerambycids and chrysomelids species of Bucharest and its surroundings, basing on bibliographical sources and the study of the collection material. A number of 365 species of superfamily Chrysomeloidea (140 cerambycids and 225 chrysomelids species), belonging to 125 genera of 16 subfamilies are listed. The species Chlorophorus herbstii, Clytus lama, Cortodera femorata, Phytoecia caerulea, Lema cyanella, Chrysolina varians, Phaedon cochleariae, Phyllotreta undulata, Cassida prasina and Cassida vittata are reported for the first time in this area. Résumé. Ce travail présente une synthèse des données concernant la présence des espèces de cerambycides et de chrysomelides de Bucarest et de ses environs, la base en étant les sources bibliographiques ainsi que l’étude du matériel existant dans les collections du musée. La liste comprend 365 espèces appartenant à la supra-famille des Chrysomeloidea (140 espèces de cerambycides et 225 espèces de chrysomelides), encadrées en 125 genres et 16 sous-familles. Les espèces Chlorophorus herbstii, Clytus lama, Cortodera femorata, Phytoecia caerulea, Lema cyanella, Chrysolina varians, Phaedon cochleariae, Phyllotreta undulata, Cassida prasina et Cassida vittata sont mentionnées pour la première fois dans cette zone Key words: Coleoptera, Chrysomeloidea, Cerambycidae, Chrysomelidae, Bucureºti (Bucharest) and surrounding areas. INTRODUCTION Data on the distribution of the cerambycids and chrysomelids species in Bucureºti (Bucharest) and the surrounding areas were published beginning with the end of the 19th century by: Jaquet (1898 a, b, 1899 a, b, 1900 a, b, 1901, 1902), Montandon (1880, 1906, 1908), Hurmuzachi (1901, 1902, 1904), Fleck (1905 a, b), Manolache (1930), Panin (1941, 1944), Eliescu et al. -

Fauna of Longicorn Beetles (Coleoptera: Cerambycidae) of Mordovia
Russian Entomol. J. 27(2): 161–177 © RUSSIAN ENTOMOLOGICAL JOURNAL, 2018 Fauna of longicorn beetles (Coleoptera: Cerambycidae) of Mordovia Ôàóíà æóêîâ-óñà÷åé (Coleoptera: Cerambycidae) Ìîðäîâèè A.B. Ruchin1, L.V. Egorov1,2 À.Á. Ðó÷èí1, Ë.Â. Åãîðîâ1,2 1 Joint Directorate of the Mordovia State Nature Reserve and National Park «Smolny», Dachny per., 4, Saransk 430011, Russia. 1 ФГБУ «Заповедная Мордовия», Дачный пер., 4, г. Саранск 430011, Россия. E-mail: [email protected] 2 State Nature Reserve «Prisursky», Lesnoi, 9, Cheboksary 428034, Russia. E-mail: [email protected] 2 ФГБУ «Государственный заповедник «Присурский», пос. Лесной, 9, г. Чебоксары 428034, Россия. KEY WORDS: Coleoptera, Cerambycidae, Russia, Mordovia, fauna. КЛЮЧЕВЫЕ СЛОВА: Coleoptera, Cerambycidae, Россия, Мордовия, фауна. ABSTRACT. This paper presents an overview of Tula [Bolshakov, Dorofeev, 2004], Yaroslavl [Vlasov, the Cerambycidae fauna in Mordovia, based on avail- 1999], Kaluga [Aleksanov, Alekseev, 2003], Samara able literature data and our own materials, collected in [Isajev, 2007] regions, Udmurt [Dedyukhin, 2007] and 2002–2017. It provides information on the distribution Chuvash [Egorov, 2005, 2006] Republics. The first in Mordovia, and some biological features for 106 survey work on the fauna of Longicorns in Mordovia species from 67 genera. From the list of fauna are Republic was published by us [Ruchin, 2008a]. There excluded Rhagium bifasciatum, Brachyta variabilis, were indicated 55 species from 37 genera, found in the Stenurella jaegeri, as their habitation in the region is region. At the same time, Ergates faber (Linnaeus, doubtful. Eight species are indicated for the republic for 1760), Anastrangalia dubia (Scopoli, 1763), Stictolep- the first time. -

The Beetle Fauna of Dominica, Lesser Antilles (Insecta: Coleoptera): Diversity and Distribution
INSECTA MUNDI, Vol. 20, No. 3-4, September-December, 2006 165 The beetle fauna of Dominica, Lesser Antilles (Insecta: Coleoptera): Diversity and distribution Stewart B. Peck Department of Biology, Carleton University, 1125 Colonel By Drive, Ottawa, Ontario K1S 5B6, Canada stewart_peck@carleton. ca Abstract. The beetle fauna of the island of Dominica is summarized. It is presently known to contain 269 genera, and 361 species (in 42 families), of which 347 are named at a species level. Of these, 62 species are endemic to the island. The other naturally occurring species number 262, and another 23 species are of such wide distribution that they have probably been accidentally introduced and distributed, at least in part, by human activities. Undoubtedly, the actual numbers of species on Dominica are many times higher than now reported. This highlights the poor level of knowledge of the beetles of Dominica and the Lesser Antilles in general. Of the species known to occur elsewhere, the largest numbers are shared with neighboring Guadeloupe (201), and then with South America (126), Puerto Rico (113), Cuba (107), and Mexico-Central America (108). The Antillean island chain probably represents the main avenue of natural overwater dispersal via intermediate stepping-stone islands. The distributional patterns of the species shared with Dominica and elsewhere in the Caribbean suggest stages in a dynamic taxon cycle of species origin, range expansion, distribution contraction, and re-speciation. Introduction windward (eastern) side (with an average of 250 mm of rain annually). Rainfall is heavy and varies season- The islands of the West Indies are increasingly ally, with the dry season from mid-January to mid- recognized as a hotspot for species biodiversity June and the rainy season from mid-June to mid- (Myers et al. -

(Coleoptera) of Peru Miguel A
University of Nebraska - Lincoln DigitalCommons@University of Nebraska - Lincoln Center for Systematic Entomology, Gainesville, Insecta Mundi Florida 2-29-2012 Preliminary checklist of the Cerambycidae, Disteniidae, and Vesperidae (Coleoptera) of Peru Miguel A. Monné Universidade Federal do Rio de Janeiro, [email protected] Eugenio H. Nearns University of New Mexico, [email protected] Sarah C. Carbonel Carril Universidad Nacional Mayor de San Marcos, Peru, [email protected] Ian P. Swift California State Collection of Arthropods, [email protected] Marcela L. Monné Universidade Federal do Rio de Janeiro, [email protected] Follow this and additional works at: http://digitalcommons.unl.edu/insectamundi Part of the Entomology Commons Monné, Miguel A.; Nearns, Eugenio H.; Carbonel Carril, Sarah C.; Swift, Ian P.; and Monné, Marcela L., "Preliminary checklist of the Cerambycidae, Disteniidae, and Vesperidae (Coleoptera) of Peru" (2012). Insecta Mundi. Paper 717. http://digitalcommons.unl.edu/insectamundi/717 This Article is brought to you for free and open access by the Center for Systematic Entomology, Gainesville, Florida at DigitalCommons@University of Nebraska - Lincoln. It has been accepted for inclusion in Insecta Mundi by an authorized administrator of DigitalCommons@University of Nebraska - Lincoln. INSECTA MUNDI A Journal of World Insect Systematics 0213 Preliminary checklist of the Cerambycidae, Disteniidae, and Vesperidae (Coleoptera) of Peru Miguel A. Monné Museu Nacional Universidade Federal do Rio de Janeiro Quinta da Boa Vista São Cristóvão, 20940-040 Rio de Janeiro, RJ, Brazil Eugenio H. Nearns Department of Biology Museum of Southwestern Biology University of New Mexico Albuquerque, NM 87131-0001, USA Sarah C. Carbonel Carril Departamento de Entomología Museo de Historia Natural Universidad Nacional Mayor de San Marcos Avenida Arenales 1256, Lima, Peru Ian P. -

Exploring Flat Faced Longhorn Beetles (Cerambycidae: Lamiinae) from the Reserve Forests of Dooars, West Bengal, India
Hindawi Publishing Corporation ISRN Entomology Volume 2013, Article ID 737193, 8 pages http://dx.doi.org/10.1155/2013/737193 Research Article Exploring Flat Faced Longhorn Beetles (Cerambycidae: Lamiinae) from the Reserve Forests of Dooars, West Bengal, India Sumana Saha,1 Hüseyin Özdikmen,2 Manish Kanti Biswas,3 and Dinendra Raychaudhuri4 1 Department of Zoology, Darjeeling Government College, Government of West Bengal, Darjeeling, West Bengal 734101, India 2 Gazi Universitesi,¨ Fen-Edebiyat Fakultesi,¨ Biyoloji Bol¨ um¨ u,¨ 06500 Ankara, Turkey 3 Department of Zoology, Sreegopal Banerjee College, Mogra, Hooghly, West Bengal 712148, India 4 Entomology Laboratory, Department of Zoology, University of Calcutta, 35 Ballygunge Circular Road, Kolkata, West Bengal 700019, India Correspondence should be addressed to Dinendra Raychaudhuri; [email protected] Received 25 June 2013; Accepted 7 August 2013 Academic Editors: Y. Fan and P. Simoes˜ Copyright © 2013 Sumana Saha et al. This is an open access article distributed under the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited. The present study deals with 29 lamiid species under 21 genera of Dooars, West Bengal, India. These include 4 newly recorded species, namely, Macrochenus isabellinus Aurivillius, Aesopida malasiaca Thomson, Pterolophia (Hylobrotus) lateralis Gahan and Nupserha quadrioculata (Thunberg) from India while 16 others (marked by ∙)fromthestate. 1. Introduction We (saving the second author) for nearly two decades are involved in the exploration of the long horn beetles of Subfamily Lamiinae (Cerambycidae) include members of flat the area. Present communication is one such outcome on the faced longhorn beetles that are both xylophagous and phy- lamiids dealing with 29 species under 21 genera. -

Miroshnikov, 2014: 19
Russian Entomological Society Sochi National Park ADVANCES IN STUDIES ON ASIAN CERAMBYCIDS (COLEOPTERA: CERAMBYCIDAE) Papers by Alexandr I. MIROSHNIKOV, dedicated to the memory of Dr. Judson Linsley GRESSITT Edited by Alexandr S. KONSTANTINOV, S. Adam Ślipiński & Alexey Yu. SOLODOVNIKOV KMK Scientific Press Ltd. Krasnodar – Moscow 2014 KONSTANTINOV A.S., Ślipiński s.A. & SOLODOVNIKOV A.Yu. (Eds): Advances in studies on Asian cerambycids (Coleoptera: Cerambycidae). Papers by Alexandr I. MIROSHNIKOV, dedicated to the memory of Dr. Judson Linsley GRESSITT. Krasnodar – Moscow: KMK Scientific Press Ltd. 2014. – 237 pp. Dr. Alexandr S. KONSTANTINOV Systematic Entomology Laboratory, USDA, c/o Smithsonian Institution, Washington, U.S.A. Dr. S. Adam Ślipiński CSIRO Australian National Insect Collection, Canberra, Australia Dr. Alexey Yu. SOLODOVNIKOV Natural History Museum of Denmark, University of Copenhagen, Denmark Cover design by A.I. Miroshnikov Frontispiece illustration by K.V. Makarov & A.I. Miroshnikov © A.I. Miroshnikov, 2014 © Sochi National Park, 2014 © Russian Entomological Society, 2014 © KMK Scientific Press Ltd., 2014 ISBN 978-5-87317-820-9 To the memory of Dr. Judson Linsley Gressitt (1914–1982), to the day of his forthcoming centenary, this work is being dedicated. CONTENTS From the editors ............................................................................................... 7 –8 From the author ................................................................................................ 9 A.I. MIROSHNIKOV. New genera -

New Longhorn Beetles (Coleoptera: Cerambycidae) from Serbia
Arch. Biol. Sci., Belgrade, 57 (4), 27P-28P, 2005. NEW LONGHORN BEETLES (COLEOPTERA: CERAMBYCIDAE) FROM SERBIA. Nataša Pil1 and D. Stojanović2. 1Institute for Nature Conservation of Serbia, 21000 Novi Sad, Serbia and Montenegro, 2”Fruška Gora” National Park, 21208 Sremska Kamenica, Serbia and Montenegro UDC 597.76(497.11) Since the 1980’s, longhorn beetles (Coleoptera, Cerambycidae) They feed in the central region of the cone or occasionally in have been only randomly researched in Serbia. From earlier the base of old scales. The life cycle probably last two years, years, there are very detailed publications on this insect group and pupation very likely occurs in the soil. Adults emerge in (A d a m o v i ć , 1965; M i k š i ć and G e o r g i j e v i ć , 1971; April-July, on flowers. The given species differs from the simi- 1973; M i k š i ć and K o r p i č , 1985). lar Cortodera humeralis (Schaller, 1783) in having only sparse pubescence on the pronotum and head, with glabrous median The most recent data (I l i ć , 2005) indicate the presence line, and sparse pubescence on the outer border of the eye and of 245 longhorn beetle species (Coleoptera: Cerambycidae) in base of the antennae. Serbia. Not included in the mentioned publication, the follow- ing five species should be added to the list: Cortodera discolor 3. Vadonia hirsuta (Daniel and Daniel,1891) Fairmaire, 1866; Stenopterus similatus Holzschuh, 1979; Chlo- rophorus aegyptiacus (Fabricius, 1775); Agapanthia osmanlis (New data: Mt. -

Central Sikhote-Alin
WHC Nomination Documentation File Name: 766rev.pdf UNESCO Region: EUROPE AND THE NORTH AMERICA __________________________________________________________________________________________________ SITE NAME: Central Sikhote-Alin DATE OF INSCRIPTION: 16th December 2001 STATE PARTY: RUSSIAN FEDERATION CRITERIA: N (iv) DECISION OF THE WORLD HERITAGE COMMITTEE: Excerpt from the Report of the 25th Session of the World Heritage Committee The Committee inscribed Central Sikhote-Alin on the World Heritage List under criterion (iv): Criterion (iv): The nominated area is representative of one of the world's most distinctive natural regions. The combination of glacial history, climate and relief has allowed the development of the richest and most unusual temperate forests in the world. Compared to other temperate ecosystems, the level of endemic plants and invertebrates present in the region is extraordinarily high which has resulted in unusual assemblages of plants and animals. For example, subtropical species such as tiger and Himalayan bear share the same habitat with species typical of northern taiga such as brown bear and reindeer. The site is also important for the survival of endangered species such as the scaly-sided (Chinese) merganser, Blakiston's fish-owl and the Amur tiger. This serial nomination consists of two protected areas in the Sikhote- Alin mountain range in the extreme southeast of the Russian Federation: NAME LOCATION AREA Sikhote-Alin Nature Preserve Terney District 401,428 ha Goralij Zoological Preserve Coastal zone on the Sea of Japan, N of Terney 4,749 ha The Committee encouraged the State Party to improve management of the Bikin River protected areas (Bikin Territory of Traditional Nature Use and Verkhnebikinski zakaznik) before nominating it as an extension. -

Systematic Notes on the Cerambycidae (Insecta: Coleoptera) Described from Burmese Amber
Palaeoentomology 002 (3): 215–218 ISSN 2624-2826 (print edition) https://www.mapress.com/j/pe/ Short PALAEOENTOMOLOGY Copyright © 2019 Magnolia Press Communication ISSN 2624-2834 (online edition) PE https://doi.org/10.11646/palaeoentomology.2.3.3 http://zoobank.org/urn:lsid:zoobank.org:pub:0688DAE1-3498-46AB-8DF8-274DD5A7A677 Systematic notes on the Cerambycidae (Insecta: Coleoptera) described from Burmese amber FRANCESCO VITALI Nationalmusée fir Naturgeschicht, rue Münster 25, L-2160 Luxembourg, Luxembourg. E-mail address: [email protected] Introduction Systematic palaeontology The description of a new fossil taxon presupposes the global Order Coleoptera Linnaeus, 1758 knowledge of the examined group and of the existence of Superfamily Cerambycoidea Latreille, 1802 possible sibling, mimicking or simply superficially similar Family Cerambycidae Latreille, 1802 taxa. The older the fossils are, the greater the possibility of Subfamily Prioninae Latreille, 1802 misidentification. Moreover, the knowledge of the assumed Tribe Meroscelisini Thomson, 1861 stat. nov. phylogeny and of the evolution centres of the extant taxa allows understanding the real taxonomy of new fossil Genus Qitianniu Lin & Bai, 2017 entities, giving consistency and support to the descriptions. Qitianniu zhihaoi Lin & Bai, 2017 Because of its unusual morphological characters, the recent description of Apophisandra ammytae Molino- According to its authors, Qitianniu zhihaoi is characterised Olmedo, 2017 (new genus, species and tribe) was by minute body size (4.6 mm), body slightly flattened disconcerting to most specialists in cerambycids. This taxon dorsoventrally, tarsi cryptopentamerous, eyes very large and evidently belongs to another family. coarsely facetted, last segment of palpi not tapered apically, This paper also revises the status of Qitianniu pronotum with complete lateral margin and antennae longer zhihaoi Lin & Bai, 2017 (whose systematic position inside than body (Lin & Bai, 2017). -
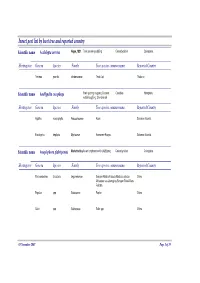
Insect Pest List by Host Tree and Reported Country
Insect pest list by host tree and reported country Scientific name Acalolepta cervina Hope, 1831 Teak canker grub|Eng Cerambycidae Coleoptera Hosting tree Genera Species Family Tree species common name Reported Country Tectona grandis Verbenaceae Teak-Jati Thailand Scientific name Amblypelta cocophaga Fruit spotting bug|eng Coconut Coreidae Hemiptera nutfall bug|Eng, Chinche del Hosting tree Genera Species Family Tree species common name Reported Country Agathis macrophylla Araucariaceae Kauri Solomon Islands Eucalyptus deglupta Myrtaceae Kamarere-Bagras Solomon Islands Scientific name Anoplophora glabripennis Motschulsky Asian longhorn beetle (ALB)|eng Cerambycidae Coleoptera Hosting tree Genera Species Family Tree species common name Reported Country Paraserianthes falcataria Leguminosae Sengon-Albizia-Falcata-Molucca albizia- China Moluccac sau-Jeungjing-Sengon-Batai-Mara- Falcata Populus spp. Salicaceae Poplar China Salix spp. Salicaceae Salix spp. China 05 November 2007 Page 1 of 35 Scientific name Aonidiella orientalis Newstead, Oriental scale|eng Diaspididae Homoptera 1894 Hosting tree Genera Species Family Tree species common name Reported Country Lovoa swynnertonii Meliaceae East African walnut Cameroon Azadirachta indica Meliaceae Melia indica-Neem Nigeria Scientific name Apethymus abdominalis Lepeletier, Tenthredinidae Hymenoptera 1823 Hosting tree Genera Species Family Tree species common name Reported Country Other Coniferous Other Coniferous Romania Scientific name Apriona germari Hope 1831 Long-horned beetle|eng Cerambycidae -

Coleoptera: Cerambycidae: Lamiinae)
INSECTA MUNDI, Vol. 16, No.4, December, 2002 247 Notes on Oriental Lamiini (Coleoptera: Cerambycidae: Lamiinae) Dr. Karl-Ernst Hudepohl Marktresidenz, Schillerstr. 8, 83209 Prien am Chiemsee, Germany Daniel J. Heffern 10531 Goldfield Ln. Houston, TX 77064 USA Longhorned beetles of the Tribe Lamiini have Vitticereopsius Breuning, 1961a:143 (type spe evolved into approximately 180 genera in South cies: Epepeotes vittipennis Fisher) is a synonym of East Asia and nearby regions. Many genera and Cereopsius Pascoe, 1862:344 (type species: Cere species are poorly-known and some taxa are still opsius exoletus Pascoe), new synonymy. Cereop undescribed. As a prelude to a checklist and further siusvittipennis (Fisher, 1935:599) becomes a new studies of this group, the authors propose some combination. taxonomicchangesand provide correctionsto previ Cereopsius sexmaculatus immaculithorax Bre ous literature. uning, 1974:238is not considered a valid subspecies The private collection and literature of the se- . of CereopsiussexmaculatusAurivillius, 1907:108, nior authorare depositedin Zoologische Staatssam since it is just a species with variable markings, mlung Munchen, MiinchhausenstraBe 21, D-8000, new synonymy. Munchen, Germany. Breuning (1968) placed Cyriepepeotes Breun ing, 1963: 17 (type species: Cyriepepeotes grossepunc Elongatorsidis Breuning, 1967a:183 (type spe tatus Breuning) as a synonym of Crucihammus cies: Elongatorsidis brunneus Breuning) is a syn Breuning, 1936:295 (type species: Crucihammus onym of Agniohammus Breuning, 1936:303 (type subcruciatus Breuning). Subsequently, inthemono species: Agniohammus olivaceus Breuning), new graph on Laotian Lamiinae (Rondon and Breuning, synonymy.Agniohammusbrunneus (Breuning, 1970), this generic synonymy is not listed, but the 1967a:183) becomes a new combination. appropriate combination of Crucihammus Perihammus Aurivillius, 1923:457 (type spe grossepunctatus Breuning as a Laotian species is cies: Perihammus bifasciatus Aurivillius) and Par given.