6Th International Conference on Thalassemia
Total Page:16
File Type:pdf, Size:1020Kb

Load more
Recommended publications
-

2018-19 Annual Report वािष�क �ितवेदन
2018-19 ANNUAL REPORT वािषक ितवेदन MAULANA AZAD NATIONAL INSTITUTE OF TECHNOLOGY, BHOPAL 462 003 मौलाना आजाद राीय ौोिगकी संथान, भोपाल 462 003 Contents Page No. Vision and Mission Statement i Organisation and Administration ii Introduction ii Board of Governors iii Finance Committee iv Building Works Committee iv Senate Members v Administrative Heads vii Heads of Department/Centre vii Chairman of various Committees vii Administrative Officers viii Director’s Note ix Academic Programmes and Department Profile 1 Academic Programmes 1 Department Profile 1 Department of Architecture and Planning 2 Department of Biological Science and Engineering 8 Department of Chemical Engineering 10 Department of Chemistry 13 Department of Civil Engineering 15 Department of Computer Science and Engineering 36 Department of Electrical Engineering 44 Department of Electronics Communication and Engineering 51 Energy Centre 58 Department of Humanities 62 Department of Management Studies 64 Department of Materials and Metallurgical Engineering 66 Department of Mathematics, Bioinformatics and Computer Applications 68 Department of Mechanical Engineering 75 Department of Physics 85 Summary of all Departments 88 Academic Programme Administration 89 Admission Statistics 89 Undergraduate 89 Post Graduate 90 Ph.D. 92 Convocation 94 Awards and Medals 96 Training and Placement 97 Students’ Activities 100 Infrastructure Facilities 103 Statement of Accounts 105 MANIT CELEBRATING 60 YEARS OF EDUCATION AND EMPOWERMENT VISION “MANIT looks forward to becoming a global centre for technical and professional knowledge” MISSION STATEMENT “To produce technical professionals abreast with competence, logical mind set, moral & ethical values and inner strength synchronous with the futuristic requirement of global business so as to strengthen the national economy” i ORGANISATION AND ADMINISTRATION ORGANISATION AND ADMINISTRATION Introduction Maulana Azad National Institute of Technology (MANIT) is one of the leading institutions of national importance in the area of technical education. -
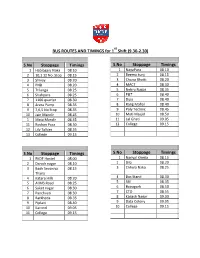
BUS ROUTES and TIMINGS for 1 Shift (9.30-2.30)
st BUS ROUTES AND TIMINGS for 1 Shift (9.30-2.30) S.No Stoppage Timings S.No Stoppage Timings 1 Habibganj Naka 08.10 1 NayaPura 08.10 2 10,1 12 No. Stop 08.15 2 Beema kunj 08.15 3 Shivay 08.20 3 Chuna Bhatti 08.20 4 PNB 08.20 4 MACT 08.30 5 Trilanga 08.25 5 Nehru Nagar 08.35 6 Shahpura 08.25 6 P&T 08.40 7 1100 quarter 08.30 7 Dipo 08.40 8 Arera Pump 08.35 8 Rang Mahal 08.40 9 7,6,5 No Stop 08.35 9 Poly Technic 08.45 10 Jain Mandir 08.45 10 Moti Masjid 08.50 1 Mata Mandir 08.45 11 Lal Ghati 09.05 11 Roshan Pura 08.50 12 College 09.15 12 Lily Talkies 08.55 13 College 09.15 S.No Stoppage Timings S.No Stoppage Timings 1 RKDF Hostel 08.00 1 Nariyal Kheda 08.15 2 Danish nagar 08.10 2 DIG 08.20 3 Bagh Sevaniya 08.15 3 Chhola Naka 08.25 Thana 4 Katara Hills 08.20 4 Bus Stand 08.30 5 AIIMS Road 08.25 5 SBI 08.35 6 Saket nagar 08.30 6 Bairagarh 08.50 7 Panchvati 08.30 7 CTO 08.55 8 Barkheda 08.35 8 Kailash Nagar 09.00 9 Piplani 08.40 9 Data Colony 09.05 10 Karond 09.05 10 College 09.15 11 College 09.15 S.No Stoppage Timings S.No Stoppage Timings 1 S P 08.00 1 IndraPuri 08.10 2 S P S 08.10 2 Beema Hospital 08.10 3 Saket Nagar 08.15 3 JK Road 08.15 4 Awadh Puri 08.25 4 Apsara 08.25 5 Vidhya Sagar 08.30 5 Pushpa Nagar 08.35 6 Piplani 08.35 6 Railway Station 08.45 7 Karond 09.10 7 Bhopal Talkies 08.45 8 College 09.15 8 Housing Board 08.50 9 Karond 09.00 10 College 09.15 S.No Stoppage Timings S.No Stoppage Timings 1 Prabhat 08.20 1 Patel Nagar 08.30 2 Ashoka Garden 08.25 2 Ratnagiri 08.35 3 Pushpa Nagar 08.35 3 Vrindavan Nagar 08.40 4 Dwarka Nagar 08.40 -

District Welfare Office,Khagaria Bc-Ebc Post Matric Scholarship Reject in Distisct 2016-17 Sl.N O
DISTRICT WELFARE OFFICE,KHAGARIA BC-EBC POST MATRIC SCHOLARSHIP REJECT IN DISTISCT 2016-17 SL.N O. COLLEGE NAME STUDENT NAME Father's Name RESON 1 2 3 4 5 B.Ed TEACHER TRAINING SUNIL KUMAR RAM PRAVESH YADAV uohu vkosnuA 1 COLLEGE ,KHAGARIA B.Ed TEACHER TRAINING PRASHANT KUMAR ASHOK KUMAR YADAV uohu vkosnuA 2 COLLEGE ,KHAGARIA B.Ed TEACHER TRAINING VIKRANT KUMAR CHANDRACHUR PRASAD uohu vkosnuA 3 COLLEGE ,KHAGARIA B.Ed TEACHER TRAINING PRIYAJEET KUMAR SUBODH KUMAR uohu vkosnuA 4 COLLEGE ,KHAGARIA B.Ed TEACHER TRAINING SUDHA KUMARI SHREE PULKIT PARSAD SINGH uohu vkosnuA 5 COLLEGE ,KHAGARIA B.Ed TEACHER TRAINING MOHIT KUMAR JAYKISHOR PRASAD YADAV uohu vkosnuA 6 COLLEGE ,KHAGARIA B.Ed TEACHER TRAINING SANTOSH KUMAR RAMCHANDRA SINGH uohu vkosnuA 7 COLLEGE ,KHAGARIA B.Ed TEACHER TRAINING RUBY KUMARI JAGARNATH YADAV uohu vkosnuA 8 COLLEGE ,KHAGARIA B.Ed TEACHER TRAINING MAMTA KUMARI DIWAKAR MEHTA uohu vkosnuA 9 COLLEGE ,KHAGARIA B.Ed TEACHER TRAINING MUKESH KUMAR MADHUKAR SRI UPENDRA PRASAD SINGH uohu vkosnuA 10 COLLEGE ,KHAGARIA B.Ed TEACHER TRAINING SINDHU KUMARI SHRI CHHOTELAL MODI uohu vkosnuA 11 COLLEGE ,KHAGARIA B.Ed TEACHER TRAINING SUSHMA KUMARI MAHENDRA YADAV uohu vkosnuA 12 COLLEGE ,KHAGARIA B.Ed TEACHER TRAINING KARMVIR KUMAR DINESH PRASAD YADAV uohu vkosnuA 13 COLLEGE ,KHAGARIA B.Ed TEACHER TRAINING PAMMI KUMARI MADAN KUMAR YADAV uohu vkosnuA 14 COLLEGE ,KHAGARIA B.Ed TEACHER TRAINING KISHOR KUMAR KRISHANA DEO VERMA uohu vkosnuA 15 COLLEGE ,KHAGARIA B.Ed TEACHER TRAINING RAJEEV KUMAR KULDEO PRASAD SAH uohu vkosnuA 16 COLLEGE ,KHAGARIA -

Consolidated List Private Universities
UNIVERSITY GRANTS COMMISSION State-wise List of Private Universities as on 06.08.2021 S.No Name of Private University Date of Notification ARUNACHAL PRADESH 1. Apex Professional University, Pasighat, District East Siang, 10.05.2013 Arunachal Pradesh - 791102. 2. Arunachal University of Studies, NH-52, Namsai, Distt – Namsai 26.05.2012 - 792103, Arunachal Pradesh. 3. Arunodaya University, E-Sector, Nirjuli, Itanagar, Distt. Papum 21.10.2014 Pare, Arunachal Pradesh-791109 4. Himalayan University, 401, Takar Complex, Naharlagun, 03.05.2013 Itanagar, Distt – Papumpare – 791110, Arunachal Pradesh. 5. North East Frontier Technical University, Sibu-Puyi, Aalo 03.09.2014 (PO), West Siang (Distt.), Arunachal Pradesh –791001. 6. The Global University, Hollongi, Itanagar, Arunachal Pradesh. 18.09.2017 7. The Indira Gandhi Technological & Medical Sciences University, 26.05.2012 Ziro, Arunachal Pradesh. 8. Venkateshwara Open University, Itanagar, Arunachal Pradesh. 20.06.2012 Andhra Pradesh 9. Bharatiya Engineering Science and Technology Innovation 17.02.2019 University, Gownivaripalli, Gorantla Mandal, Anantapur, Andhra Pradesh 10. Centurian University of Technology and Management, Gidijala 23.05.2017 Junction, Anandpuram Mandal, Visakhapatnam- 531173, Andhra Pradesh. 11. KREA University, 5655, Central, Expressway, Sri City-517646, 30.04.2018 Andhra Pradesh 12. Saveetha Amaravati University, 3rd Floor, Vaishnavi Complex, 30.04.2018 Opposite Executive Club, Vijayawada- 520008, Andhra Pradesh 13. SRM University, Neerukonda-Kuragallu Village, mangalagiri 23.05.2017 Mandal, Guntur, Dist- 522502, Andhra Pradesh (Private University) 14. VIT-AP University, Amaravati- 522237, Andhra Pradesh (Private 23.05.2017 University) ASSAM 15. Assam Don Bosco University, Azara, Guwahati 12.02.2009 16. Assam Down Town University, Sankar Madhab Path, Gandhi 29.04.2010 Nagar, Panikhaiti, Guwahati – 781 036. -

List of Approved Institutes in 2015-16
List of Approved Institutes in 2015-16 Approved Sr. Application P / F Level of NRI PIO Foreign State Region Institute Name Program Shift Course Intake 15- University Name No. Number Time Course Approved Approved Approved 16 SRI PARASHURAM INSTITUTE ENGINEERING Madhya 1st FULL UNDER Rajiv Gandhi Prodoyogiki 1 1-2450403601 Central OF TECHNOLOGY AND AND CIVIL ENGINEERING 60 NA NA NA Pradesh Shift TIME GRADUATE Vishwavidyalaya, Bhopal RESEARCH TECHNOLOGY SRI PARASHURAM INSTITUTE ENGINEERING Madhya 1st FULL UNDER COMPUTER SCIENCE & Rajiv Gandhi Prodoyogiki 2 1-2450403601 Central OF TECHNOLOGY AND AND 60 NA NA NA Pradesh Shift TIME GRADUATE ENGINEERING Vishwavidyalaya, Bhopal RESEARCH TECHNOLOGY SRI PARASHURAM INSTITUTE ENGINEERING ELECTRICAL AND Madhya 1st FULL UNDER Rajiv Gandhi Prodoyogiki 3 1-2450403601 Central OF TECHNOLOGY AND AND ELECTRONICS 60 NA NA NA Pradesh Shift TIME GRADUATE Vishwavidyalaya, Bhopal RESEARCH TECHNOLOGY ENGINEERING ELECTRONICS AND SRI PARASHURAM INSTITUTE ENGINEERING Madhya 1st FULL UNDER COMMUNICATION Rajiv Gandhi Prodoyogiki 4 1-2450403601 Central OF TECHNOLOGY AND AND 60 NA NA NA Pradesh Shift TIME GRADUATE ENGINEERING Vishwavidyalaya, Bhopal RESEARCH TECHNOLOGY (SANDWICH) SRI PARASHURAM INSTITUTE ENGINEERING Madhya 1st FULL UNDER MECHANICAL Rajiv Gandhi Prodoyogiki 5 1-2450403601 Central OF TECHNOLOGY AND AND 60 NA NA NA Pradesh Shift TIME GRADUATE ENGINEERING Vishwavidyalaya, Bhopal RESEARCH TECHNOLOGY ENGINEERING Madhya MILLENNIUM INSTITUTE OF 1st FULL POST COMPUTER SCIENCE Rajiv Gandhi Prodoyogiki 6 1-2450499150 -
![=V >` UV De`\Vd CRWR]V S]Rkv](https://docslib.b-cdn.net/cover/2895/v-uv-de-vd-crwr-v-s-rkv-2302895.webp)
=V >` UV De`\Vd CRWR]V S]Rkv
C M Y K ( D 4+ #E #E E +,-+ &./01 RNI Regn. No. MPENG/2004/13703, Regd. No. L-2/BPLON/41/2006-2008 @% ) ()* 0A++' 0?0 @7 :7 '+%$5 ,' !4 &' 23 &$'7((:&'( :( 5! 5:5 %75,)<: (77<7,*:7 $% &):'(7 :<*7;7 7%%$7,;7%7:C<$*$$', 5 *7$$,4$7, $! # .1!2.34 /51> ! "$% !& ;7+7,47=5+7 R !" # $ !% $ ,:;4:%<$ the authorities and upon fur- panies operating in France. ! " ther investigation for the peri- A spokesperson of Reliance # fresh controversy broke od 2010 to 2012 an additional Communications said here the Aout with a French news- tax of 91 mn was levied. tax demands were “complete- paper report that industrialist The total tax liability as per ly unsustainable and illegal” Anil Ambani’s firm was grant- the French newspaper was 151 and that the company denied Q ed tax waiver worth 143.7 mil- million Euros and six months any favouritism or gain from $% & " lion Euros months after India after the announcement by the settlement. “During the &$ announced the decision to buy India to buy Rafale jets, the period under consideration by ' &$' Rafale fighter jets. The Centre French tax authorities made a the French Tax Authorities $% && # $ moved swiftly and termed it as settlement with the company 2008-2012 i.e. nearly 10 years ( )*" % a conjectural mischievous for 7.3 million Euros instead of ago, Flag France had an oper- # Q attempt to disinform and said 151 million Euros. ating loss of 20 crore (Euro 2.7 &''( )* any connection with the Rafale Reacting to the article, million). French tax authorities ! + , - issue was totally inaccurate. -

List of Universities Under Section 2(F) and Section 3 of the UGC Act, 1956
UNIVERSITY GRANTS COMMISSION List of Universities under Section 2(f) and Section 3 of the UGC Act, 1956 S.No ANDHRA PRADESH 1. A.P. University of Law, Palace Layout, Pedawaltair, Visakhapatnam – 530 017 (A. P) (State University). 2. Acharya N.G. Ranga Agricultural University, Hderabad-500 030. (State University) 3. Acharya Nagarjuna University, Nagarjuna Nagar, Guntur-522 510. (State University) 4. Adikavi Nannaya University, Jaya Krishnapuram, Rajahmundry – 533 105, Andhra Pradesh. (State University) 5. Andhra Pradesh University of Health Sciences, Vijayawada-520 008. (State University) 6. Andhra University, Visakhapatnam-530 003. (State University) 7. Dr. B.R. Ambedkar Open University, Jubilee Hills, Hyderabad-500 033. (State University) 8. Dr. B.R. Ambedkar University, Etcherla – 532 410 Srikakulam, Andhra Pradesh (State University) 9. Dravidian University, Kuppam-517 425. (State University) 10. Dr. Y.S.R. Horticultural University, PO Box No. 7, Venkataramannagudem, West Godavari District – 536 101, Andhra Pradesh. (State University) 11. Gandhi Institute of Technology and Management (GITAM), Gandhi Nagar Campus, Rushikonda, Visakhapatman – 530 045.(Deemed University) 12. ICFAI Foundation for Higher Education, Hyderabad, Andhra Pradesh. (Deemed University) 13. International Institute of Information Technology, Hyderabad-500 019. (Deemed University) 14. Jawaharlal Nehru Architecture and Fine Arts University, Mahaveer Marg, Masab Tank, Hyderabad – 500 028 (State University) 15. Jawaharlal Nehru Technological University, Anantpur-515 002, Andhra Pradesh (State University) 16. Jawaharlal Nehru Technological University, Hyderabad-500 072. (State University) 17. Jawaharlal Nehru Technological University, Kakinada-533003, Andhra Pradesh.(State University) 18. Kakatiya University, Warangal-506 009. (State University) 19. Koneru Lakshmaiah Education Foundation, Greenfields, Kunchanapalli Post, Vaddeswaram, Guntur District, Andhra Pradesh (Deemed University) 20. -

Decision of 100Th CC (14Th & 15Th November, 2016) Regarding
F 1 Decision of 100th CC (14th & 15th November, 2016) regarding approval of pharmacy institutions* * Consideration of approval of the Diploma, Degree, Pharm.D and Pharm.D (Post Baccalaureate), M.Pharm and B.Pharm (Practice) course and examination in Pharmacy. ----------------------------------------------------------------------------------------------------------------------------- A. For the approval u/s 12 "(1) In pursuance of the provisions of sub section (1) of section 12 of the Pharmacy Act, 1948 (8 of 1948), the Pharmacy Council of India declares the course in Pharmacy (as applicable) conducted by institutions mentioned below to be an approved course of study for the purpose of admission to an approved examination in Pharmacy (as applicable) in respect of number of students and academic session as specified here under: (2) In pursuance of the provisions of sub-section (2) of section 12 of the Pharmacy Act, 1948 (8 of 1948), the Pharmacy Council of India declares the Examination in Pharmacy (as applicable) held by the Examining Authorities mentioned against their name, during the session mentioned to be an approved examination for the purpose of qualifying for registration as a Pharmacist under the said Act.” B. The approval is subject to - a) the institution shall submit SIF every year as per the Time-Schedule prescribed by the Council. b) the institution shall submit annual affiliation fee on or before due date. c) the institution shall appoint the teaching faculty with the qualification and experience as prescribed under the “Minimum Qualification for Teachers in Pharmacy Institutions Regulations, 2014”. C. Besides above conditions, institutions seeking approval of Pharm.D / Pharm.D (Post Baccalaureate) course shall comply with the following conditions - 1. -

RAM KRISHAN DHARMARTH FOUNDATION UNIVERSITY (Gandhinagar Campus, Bhopal)
RAM KRISHAN DHARMARTH FOUNDATION UNIVERSITY (Gandhinagar campus, Bhopal) ADMISSION GUIDELINES FOR SESSION 2020-21 The information about the courses offered under various Constituent Institutttions and the Teaching Departments of the University alongwith the details like admission rules, seat intake, minimum eligibility criteria for admission, basis of seat allotment is listed course-wise in the attached Table (Annexure-1). These details are also displayed on the University Website www.rkdf.ac.in Process of counseling and admissions starts from 1st April 2020 and continues till last date of admissions. It will be held on every working day of University on the basis of merit marks obtained in the entrance examination (if require) or on the basis of the marks obtained in qualifying examination as mentioned in the Annexure -1 ****After approval in case of lockdown following admission procedure will be considered: All admission procedure will be conducted through online link www.rkdf.ac.in/admissionform.php After lockdown students must submit all original documents for verification to counseling department. Original TC/Leaving Certificate, Character Certificate and Migration Certificate will be submitted in the allotted constituent institutions or teaching departments. Seats allotment in courses will be done as mentioned in Annexure-1 i.e. first on the basis of marks obtained in entrance examination (if required) and thereafter for all the remaining seats on the basis of marks obtained in the qualifying examination. Seats allotment for reserved category will de done as per the ordinance of RKDF University which comprises of respective Regulatory Bodies/ Govt. of Madhya Pradesh directives. Applicant shall ensure the requirement of minimum educational qualification eligibility and age (if required) (As per Annexure -1) at the time of application submission. -
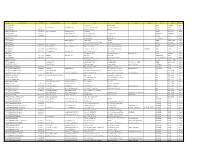
Sr CENTRE NAME TELEPHONE NO BUILDING NAME EMAIL ID ADD 1 ADD 2 ADD 3 ADD 4 DIST STATE PIN CODE
Sr CENTRE NAME TELEPHONE NO BUILDING NAME EMAIL ID ADD 1 ADD 2 ADD 3 ADD 4 DIST STATE PIN CODE 0001 ABHANPUR Shri G Colony Behind Bus Stand Raipur Chhattisgarh 493661 0002 ABOHAR 01634222060 Rajyoga Bhawan [email protected] H No.: 379, Old Fazilka Road Opp. Sham Bihar Colony Fazilka Punjab 152116 0003 ACHALDA 05681234070 H/o: Shyamdas Sarrof Nahar Bazzar Auraiya Uttar Pradesh 206241 0004 ACHALPUR CAMP 07223221353 Shiv Darshan Bhawan [email protected] Mohan Nagar Amravati Maharashtra 444805 0005 ACHAMPET 08541272089 [email protected] 19/182/3, Maruti Nagar Opp. Market Yard Mahabubnagar TELANGANA 509375 0006 ADAMPUR DOABA 01812753999 [email protected] Behind High Cross Mall Ward No. :8 Jalandhar Punjab 144102 0007 ADAPUR H/o: Muna Singh Belwa, Ward No: 12 East Champaran Bihar 845301 0008 ADASPUR [email protected] Medial Chhaka Tal: Niali Cuttack Odisha (Orissa) 754011 0009 ADAVAD H No.: 1492/2, Near Hareshwar Temple Teh: Chopda Jalgaon Maharashtra 425303 0010 ADDANKI 08593223555 Divyanubhuti Bhawan H.No. 30-210, Near Junior College Satyanarayan Kalamandir Street Prakasam Andhra Pradesh 523201 0011 ADILABAD 08732231028 SUKH SAROVAR [email protected] Near S. P. Camp Office Beside D W M A, Kailash Nagar Adilabad TELANGANA 504001 0012 ADIPUR 02836257577 Shiv Kalyankari Bhawan Plot No-C.C.3, Ward No-6 B Opp-Learner Accodemy School Gandhidham Kutch Gujarat 370201 0013 ADONI 08512251887 Paramjyoti Bhawan [email protected] Near Y.M.K. High School Mahaveer Colony Kurnool Andhra Pradesh 518301 0014 ADRA H/o: Subhankar Mitra Near By Dvc More (Chowk) Kanta Raguni Colony Purulia West Bengal 723121 0015 ADUR 04734224676 Siddharth [email protected] Opp. -

1 EPCO Institute of Environmental Studies
1 ANNEXURE List of Dissertation/ Short term Project submitted by PGDEM Students at EIES I. 2016-17 S. No Title Institute / College / University 1 Report on Solid Waste Management Mr. G.K. Mishra, EIA Expert / FAE for Solid & Hazardous Waste, M/s In Situ Envirocare, Submitted by Abhishek Mohan Bhopal 2 Habitat conservation Submitted by Anshul Dr. M.K. Singh, IIFM, Bhopal Upadhyay 3 Grid connected roof top domestic solar lighting Mr. Surendra Bajpai, Executive Engineer, Urja Submitted by Archana Pandey Vikas Nigam, Bhopal 4 Soil stabilization using waste fiber materials Dr. R. Singh, Professor, Deptt. Of Civil Submitted by Ashish kant Ahirwar Engineering, Ujjain Engineering College, Ujjain (MP) 5 A project report on Optimization of Poly Dr. Preeti Jain, AGM (QC), Grasim Industries Aluminium Chloride (PAC) in Water Treatment Ltd., Nagda Plant Submitted by Atul Sharma 6 Water pollution Dr. Rekha Shrivastava, Professor & Head, Deptt. Of Zoology, Govt. M.V.M., Bhopal Submitted by Balwan Singh Kanoje 7 Jhansi Cantonment Board Solid Waste Mr. M.K. Sharma, Envt. Suprintendent, Chawni Management Submitted by Bhanu Sharma Parishad, Jhansi 8 Kotra Sewage Treatment Plant, Bhopal Dr. Vinita Vipat, CSO, EPCO, Bhopal Submitted by Bhartesh vaibhav Jain 9 Comprehensive Environmental Pollution Index Mr. P.R. Deo (SSO) & RO, MPPCB, Singrauli (CEPI) (MP) Submitted by Brijendra kumar Gupta 10 Scenario of MSW Management, its processing, Dr. Akhilesh Upadhayay, Health Officer, environmental effects and some suggestions Indore Nagar Nigam, Indore Submitted by Deepankar Mandloi 11 Environmental Planning on Green Space (A case Dr. Dilip Thakur, Prof. & HOD Applied of new Bhopal) Science, SRT, People's University, Bhanpur, Bhopal-37 Submitted by Govind singh Solanki 12 Monitoring of air quality and pollution status of Dr. -
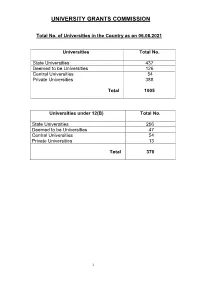
Consolidated List of All Universities
UNIVERSITY GRANTS COMMISSION Total No. of Universities in the Country as on 06.08.2021 Universities Total No. State Universities 437 Deemed to be Universities 126 Central Universities 54 Private Universities 388 Total 1005 Universities under 12(B) Total No. State Universities 256 Deemed to be Universities 47 Central Universities 54 Private Universities 13 Total 370 1 S.No ANDHRA PRADESH Date/Year of Notification/ Establishment 1. Acharya N.G. Ranga Agricultural University, Lam, 1964 Guntur – 522 034,Andhra Pradesh (State University) 2. Acharya Nagarjuna University, Nagarjuna Nagar-522510, Dt. Guntur, 1976 Andhra Pradesh. (State University) 3. Adikavi Nannaya University, 25-7-9/1, Jayakrishnapuram, 2006 Rajahmundry – 533 105, East Godavari District, Andhra Pradesh. (State University) 4. Andhra University, Waltair, Visakhapatnam-530 003, Andhra Pradesh. 1926 (State University) 5. Bharatiya Engineering Science and Technology Innovation University, 17.02.2019 Gownivaripalli, Gorantla Mandal, Anantapur, Andhra Pradesh (Private University) 6. Central University of Andhra Pradesh, IT Incubation Centre Building, 05.08.2019 JNTU Campus, Chinmaynagar, Anantapuramu, Andhra Pradesh- 515002 (Central University) 7. Central Tribal University of Andhra Pradesh, Kondakarakam, 05.08.2019 Vizianagaram, Andhra Pradesh 535008 (Central University) 8. Centurion University of Technology and Management, Gidijala Junction, 23.05.2017 Anandapuram Mandal, Visakhapatnam – 531173, Andhra Pradesh. (Private University) 9. Damodaram Sanjivayya National Law University, Plot No. 116, Sector 2008 11 MVP Colony, Visakhapatnam – 530 017, Andhra Pradesh. (State University) 10. Dr. Abdul Haq Urdu University, Kurnool- 518001, Andhra Pradesh 14.12.2018 (State University) 11. Dr. B.R. Ambedkar University, Etcherla, Dt. Srikakulam-532410, 2008 Andhra Pradesh. (State University) 12. Dravidian University, Srinivasanam, -517 425, Chittoor District, 1997 Andhra Pradesh.