Clinical Protocol IM101566
Total Page:16
File Type:pdf, Size:1020Kb

Load more
Recommended publications
-

COVID-19—The Potential Beneficial Therapeutic Effects of Spironolactone During SARS-Cov-2 Infection
pharmaceuticals Review COVID-19—The Potential Beneficial Therapeutic Effects of Spironolactone during SARS-CoV-2 Infection Katarzyna Kotfis 1,* , Kacper Lechowicz 1 , Sylwester Drozd˙ zal˙ 2 , Paulina Nied´zwiedzka-Rystwej 3 , Tomasz K. Wojdacz 4, Ewelina Grywalska 5 , Jowita Biernawska 6, Magda Wi´sniewska 7 and Miłosz Parczewski 8 1 Department of Anesthesiology, Intensive Therapy and Acute Intoxications, Pomeranian Medical University in Szczecin, 70-111 Szczecin, Poland; [email protected] 2 Department of Pharmacokinetics and Monitored Therapy, Pomeranian Medical University, 70-111 Szczecin, Poland; [email protected] 3 Institute of Biology, University of Szczecin, 71-412 Szczecin, Poland; [email protected] 4 Independent Clinical Epigenetics Laboratory, Pomeranian Medical University, 71-252 Szczecin, Poland; [email protected] 5 Department of Clinical Immunology and Immunotherapy, Medical University of Lublin, 20-093 Lublin, Poland; [email protected] 6 Department of Anesthesiology and Intensive Therapy, Pomeranian Medical University in Szczecin, 71-252 Szczecin, Poland; [email protected] 7 Clinical Department of Nephrology, Transplantology and Internal Medicine, Pomeranian Medical University, 70-111 Szczecin, Poland; [email protected] 8 Department of Infectious, Tropical Diseases and Immune Deficiency, Pomeranian Medical University in Szczecin, 71-455 Szczecin, Poland; [email protected] * Correspondence: katarzyna.kotfi[email protected]; Tel.: +48-91-466-11-44 Abstract: In March 2020, coronavirus disease 2019 (COVID-19) caused by SARS-CoV-2 was declared Citation: Kotfis, K.; Lechowicz, K.; a global pandemic by the World Health Organization (WHO). The clinical course of the disease is Drozd˙ zal,˙ S.; Nied´zwiedzka-Rystwej, unpredictable but may lead to severe acute respiratory infection (SARI) and pneumonia leading to P.; Wojdacz, T.K.; Grywalska, E.; acute respiratory distress syndrome (ARDS). -
Guidance for the Format and Content of the Protocol of Non-Interventional
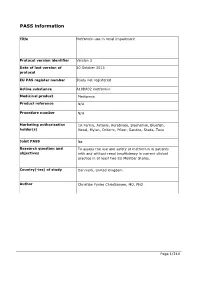
PASS information Title Metformin use in renal impairment Protocol version identifier Version 2 Date of last version of 30 October 2013 protocol EU PAS register number Study not registered Active substance A10BA02 metformin Medicinal product Metformin Product reference N/A Procedure number N/A Marketing authorisation 1A Farma, Actavis, Aurobindo, Biochemie, Bluefish, holder(s) Hexal, Mylan, Orifarm, Pfizer, Sandoz, Stada, Teva Joint PASS No Research question and To assess the use and safety of metformin in patients objectives with and without renal insufficiency in current clinical practice in at least two EU Member States. Country(-ies) of study Denmark, United Kingdom Author Christian Fynbo Christiansen, MD, PhD Page 1/214 Marketing authorisation holder(s) Marketing authorisation N/A holder(s) MAH contact person N/A Page 2/214 1. Table of Contents PASS information .......................................................................................................... 1 Marketing authorisation holder(s) .................................................................................... 2 1. Table of Contents ...................................................................................................... 3 2. List of abbreviations ................................................................................................... 4 3. Responsible parties .................................................................................................... 5 4. Abstract .................................................................................................................. -

Selection of a Mineralocorticoid Receptor Antagonist for Patients with Hypertension Or Heart Failure
European Journal of Heart Failure (2014) 16, 143–150 REVIEW doi:10.1111/ejhf.31 Selection of a mineralocorticoid receptor antagonist for patients with hypertension or heart failure Javaid Iqbal1*, Yasir Parviz1, Bertram Pitt2, John Newell-Price3, Abdallah Al-Mohammad1, and Faiez Zannad4 1Department of Cardiovascular Science at the University of Sheffield and Cardiology Department at Sheffield Teaching Hospitals NHS Trust, Sheffield,K; U 2Cardiovascular Centre, University of Michigan, Ann Arbor, MI, USA; 3Department of Human Metabolism at the University of Sheffield and Endocrinology Department at Sheffield Teaching Hospitals NHS Trust, Sheffield, UK; and 4INSERM, Centre d’Investigation Clinique and Centre Hospitalier Universitaire, and the Department of Cardiology, Nancy University, Université de Lorraine, Nancy, France Received 8 April 2013; revised 15July2013; accepted 19July2013; online publish-ahead-of-print 14 December 2013 Clinical trials have demonstrated morbidity and mortality benefits of mineralocorticoid receptor antagonists (MRAs) in patients with heart failure. These studies have used either spironolactone or eplerenone as the MRA. It is generally believed that these two agents have the same effects, and the data from studies using one drug could be extrapolated for the other. National and international guidelines do not generally discriminate between spironolactone and eplerenone, but strongly recommend using an MRA for patients with heart failure due to LV systolic dysfunction and post-infarct LV systolic dysfunction. There are no major clinical trials directly comparing the efficacy of these two drugs. This article aims to compare the pharmacokinetics and pharmacodynamics of spironolactone and eplerenone, and to analyse the available data for their cardiovascular indications and adverse effects. -

Aldactazide® 25
PRODUCT MONOGRAPH ALDACTAZIDE® 25 (spironolactone and hydrochlorothiazide tablets USP) tablets 25mg: 25mg ALDACTAZIDE® 50 (spironolactone and hydrochlorothiazide tablets USP) tablets 50mg: 50mg Aldosterone Antagonist with a Diuretic Pfizer Canada ULC Date of Revision: 17,300 Trans-Canada Highway April 26, 2019 Kirkland, Quebec H9J 2M5 ® TM G.D. Searle LLC Pfizer Canada ULC, Licensee © Pfizer Canada ULC 2019 Submission Control No: 224433 ALDACTAZIDE (spironolactone and hydrochlorothiazide) Page 1 of 38 Table of Contents PART I: HEALTH PROFESSIONAL INFORMATION ........................................................ 3 SUMMARY PRODUCT INFORMATION ....................................................................... 3 INDICATIONS AND CLINICAL USE ............................................................................. 3 CONTRAINDICATIONS .................................................................................................. 4 WARNINGS AND PRECAUTIONS ................................................................................. 5 ADVERSE REACTIONS ................................................................................................. 10 DRUG INTERACTIONS ................................................................................................. 16 DOSAGE AND ADMINISTRATION ............................................................................. 20 OVERDOSAGE ............................................................................................................... 21 ACTION AND CLINICAL PHARMACOLOGY -

Aldactone® Spironolactone Tablets, USP
NDA 12-151/S-062 Page 2 Aldactone® spironolactone tablets, USP WARNING Aldactone has been shown to be a tumorigen in chronic toxicity studies in rats (see Precautions). Aldactone should be used only in those conditions described under Indications and Usage. Unnecessary use of this drug should be avoided. DESCRIPTION Aldactone oral tablets contain 25 mg, 50 mg, or 100 mg of the aldosterone antagonist spironolactone, 17-hydroxy-7α-mercapto-3-oxo-17α-pregn-4-ene-21-carboxylic acid γ-lactone acetate, which has the following structural formula: Spironolactone is practically insoluble in water, soluble in alcohol, and freely soluble in benzene and in chloroform. Inactive ingredients include calcium sulfate, corn starch, flavor, hypromellose, iron oxide, magnesium stearate, polyethylene glycol, povidone, and titanium dioxide. ACTIONS / CLINICAL PHARMACOLOGY Mechanism of action: Aldactone (spironolactone) is a specific pharmacologic antagonist of aldosterone, acting primarily through competitive binding of receptors at the aldosterone-dependent sodium-potassium exchange site in the distal convoluted renal tubule. Aldactone causes increased amounts of sodium and water to be excreted, while potassium is retained. Aldactone acts both as a diuretic and as an antihypertensive drug by this mechanism. It may be given alone or with other diuretic agents which act more proximally in the renal tubule. Aldosterone antagonist activity: Increased levels of the mineralocorticoid, aldosterone, are present in primary and secondary hyperaldosteronism. Edematous states in which secondary aldosteronism is usually involved include congestive heart failure, hepatic cirrhosis, and the nephrotic syndrome. By competing with aldosterone for receptor sites, Aldactone provides effective therapy for the edema and ascites in those conditions. -

Progesterone and Related Progestins: Potential New Health Benefi Ts
CLIMACTERIC 2013;16(Suppl 1):69–78 Progesterone and related progestins: potential new health benefi ts R. Sitruk-Ware and M. El-Etr * Population Council and Rockefeller University, New York, NY, USA; * INSERM UMR 788, Kremlin Bic ê tre, France Key words: PROGESTERONE , PROGESTIN , NESTORONE, NEUROREGENERATION, MYELIN REPAIR, STROKE , HORMONE REPLACEMENT THERAPY, LACTATION , PROGESTERONE VAGINAL RING ABSTRACT Progesterone is a steroid hormone that is essential for the regulation of reproductive function. The main physiological roles of this hormone have been widely described. Progesterone and progestins have been approved for a number of indications including the treatment of irregular and anovulatory menstrual cycles and, when combined with estrogen, for contraception, and the prevention of endometrial hyperplasia in postmenopausal hormonal replacement therapy (HRT) regimens. Lack of understanding between the differ- ences in categories of the progestins as well as with the physiological hormone has resulted in considerable controversy surrounding the use of progestins for HRT regimens. Newer evidence suggests that there are distinct differences between the molecules and there is no progestin class effect, with regard to benefi ts or side-effects. In addition to its role in reproduction, progesterone regulates a number of biologically distinct processes in other tissues, particularly in the nervous system and the vessels. Recently, it has been shown in animal experiments that progesterone and the progestin Nestorone ® have positive effects on neuroregeneration and repair of brain damage, as well as myelin repair. The potential benefi ts of natural progesterone and its related derivatives warrant further investigation. It is hoped that a better understanding of the mechanism of action of progesterone and selected progestins will help in defi ning better therapies for men and women. -

SC-26304) (Kidney/Aldosterone Receptors/Tritiated Spirolactone) DIANA MARVER, JOHN STEWART*, JOHN W
Proc. Nat. Acad. Sci. USA Vol. 71, No. 4, pp. 1431-1435, April 1974 Renal Aldosterone Receptors: Studies with [3H]Aldosterone and the Anti-Mineralocorticoid [3H]Spirolactone (SC-26304) (kidney/aldosterone receptors/tritiated spirolactone) DIANA MARVER, JOHN STEWART*, JOHN W. FUNDERt, DAVID FELDMANT, AND ISIDORE S. EDELMAN Cardiovascular Research Institute and the Departments of Medicine, and of Biochemistry and Biophysics, University of California, San Francisco, Calif. 94143 Contributed by Isidre S. Edelman, January 30, 1974 ABSTRACT In vivo, a spirolactone (SC-26304) in- sterone binding site, to explore the characteristics of the aldo- hibited the effects of aldosterone on urinary K+: Na+ ratios sterone and and the binding of ['Hialdosterone to renal cytoplasmic receptor system the mechanism of the inhibitory and nuclear receptors. Cytoplasmic binding of ['flaldo_ effect (8). sterone and ['Hispirolactone (SC-26304) was similar in mag- nitude and involved the same set of sites. Under three MATERIALS AND METHODS sets of conditions-(i) in the intact rat, (ii) in kidney slices, and (iii) in reconstitution studies (mixing pre- Materials. 1,2-d-['H]Aldosterone (54 Ci/mmol) was ob- labeled cytoplasm with either purified renal nuclei or tained from New England Nuclear Corp., Boston, Mass. chromatin), ['Hispirolactone (SC-26304) did not yield [3H]SC-26304 (16 Ci/mmol) and unlabeled SC-26304 were specific nuclear complexes in contrast to the reproducible from the G. D. Searle generation of these complexes with [Hilaldosterone. In generous gifts Co., Chicago, Ill. Low glycerol density gradients, cytoplasmic ['Hialdosterone K+ food pellets (1.2 Aeq. of K+/g) were obtained from General receptor complexes sedimented at 8.5 S and 4 S in low Biochemical, Chagrin Falls, Ohio. -

ALDACTAZIDE- Spironolactone and Hydrochlorothiazide Tablet, Film Coated
ALDACTAZIDE- spironolactone and hydrochlorothiazide tablet, film coated Pfizer Laboratories Div Pfizer Inc ---------- Aldactazide® spironolactone and hydrochlorothiazide tablets DESCRIPTION ALDACTAZIDE oral tablets contain: spironolactone . 25 mg hydrochlorothiazide . 25 mg or spironolactone . 50 mg hydrochlorothiazide . 50 mg Spironolactone (ALDACTONE®), an aldosterone antagonist, is 17-hydroxy-7α-mercapto- 3-oxo-17α-pregn-4-ene-21-carboxylic acid γ-lactone acetate and has the following structural formula: Spironolactone is practically insoluble in water, soluble in alcohol, and freely soluble in benzene and in chloroform. Hydrochlorothiazide, a diuretic and antihypertensive, is 6-chloro-3,4-dihydro-2H-1,2,4- benzothiadiazine-7-sulfonamide 1,1-dioxide and has the following structural formula: Hydrochlorothiazide is slightly soluble in water and freely soluble in sodium hydroxide solution. Inactive ingredients include calcium sulfate, corn starch, flavor, hydroxypropyl cellulose, hypromellose, iron oxide, magnesium stearate, polyethylene glycol, povidone, and titanium dioxide. ACTIONS / CLINICAL PHARMACOLOGY Mechanism of action ALDACTAZIDE is a combination of two diuretic agents with different but complementary mechanisms and sites of action, thereby providing additive diuretic and antihypertensive effects. Additionally, the spironolactone component helps to minimize the potassium loss characteristically induced by the thiazide component. The diuretic effect of spironolactone is mediated through its action as a specific pharmacologic antagonist of aldosterone, primarily by competitive binding of receptors at the aldosterone-dependent sodium-potassium exchange site in the distal convoluted renal tubule. Hydrochlorothiazide promotes the excretion of sodium and water primarily by inhibiting their reabsorption in the cortical diluting segment of the distal renal tubule. ALDACTAZIDE is effective in significantly lowering the systolic and diastolic blood pressure in many patients with essential hypertension, even when aldosterone secretion is within normal limits. -

Downloaded from Bioscientifica.Com at 10/01/2021 06:44:53AM Via Free Access
57 1 S GIATTI and others Progestins in the brain 57: 2 R109–R126 Review The other side of progestins: effects in the brain Correspondence Silvia Giatti, Roberto Cosimo Melcangi and Marzia Pesaresi should be addressed Department of Pharmacological and Biomolecular Sciences, Center of Excellence on Neurodegenerative to R C Melcangi Diseases, Università degli Studi di Milano, Milan, Italy Email [email protected] Abstract Progestins are a broad class of progestational agents widely differing in their Key Words chemical structures and pharmacological properties. Despite emerging data f progesterone suggest that progestins, besides their action as endometrial protection, can also have f testosterone multiple nonreproductive functions, much remains to be discovered regarding the f combined oral actions exerted by these molecules in the nervous system. Here, we report the role contraceptive exerted by different progestins, currently used for contraception or in postmenopausal f hormone replacement therapy hormone replacement therapies, in regulating cognitive functions as well as social f neuroprotection behavior and mood. We provide evidence that the effects and mechanisms underlying their actions are still confusing due to the use of different estrogens and progestins as well as different doses, duration of exposure, route of administration, baseline hormonal status and age of treated women. We also discuss the emerging issue concerning the relevant increase of these substances in the environment, able to deeply affect aquatic wildlife as well as to exert a possible influence in humans, which may be exposed to these compounds via contaminated drinking water and seafood. Journal of Molecular Endocrinology Finally, we report literature data showing the neurobiological action of progestins and in particular their importance during neurodegenerative events. -

ALDACTONE (Spironolactone)
Aldactone® spironolactone tablets, USP WARNING ALDACTONE has been shown to be a tumorigen in chronic toxicity studies in rats (see Precautions). ALDACTONE should be used only in those conditions described under Indications and Usage. Unnecessary use of this drug should be avoided. DESCRIPTION ALDACTONE oral tablets contain 25 mg, 50 mg, or 100 mg of the aldosterone antagonist spironolactone, 17-hydroxy-7α-mercapto-3-oxo-17α-pregn-4-ene-21- carboxylic acid γ-lactone acetate, which has the following structural formula: Spironolactone is practically insoluble in water, soluble in alcohol, and freely soluble in benzene and in chloroform. Inactive ingredients include calcium sulfate, corn starch, flavor, hypromellose, iron oxide, magnesium stearate, polyethylene glycol, povidone, and titanium dioxide. ACTIONS / CLINICAL PHARMACOLOGY Mechanism of action: ALDACTONE (spironolactone) is a specific pharmacologic antagonist of aldosterone, acting primarily through competitive binding of receptors at the aldosterone-dependent sodium-potassium exchange site in the distal convoluted renal tubule. ALDACTONE causes increased amounts of sodium and water to be excreted, while potassium is retained. ALDACTONE acts both as a diuretic and as an antihypertensive drug by this mechanism. It may be given alone or with other diuretic agents that act more proximally in the renal tubule. Aldosterone antagonist activity: Increased levels of the mineralocorticoid, aldosterone, are present in primary and secondary hyperaldosteronism. Edematous states in which secondary aldosteronism is usually involved include congestive heart failure, hepatic cirrhosis, and nephrotic syndrome. By competing with aldosterone for receptor sites, ALDACTONE provides effective therapy for the edema and ascites in those conditions. ALDACTONE counteracts secondary aldosteronism induced by the volume depletion and associated sodium loss caused by active diuretic therapy. -

Spironolactone 25 Mg
1. NAME OF THE MEDICINAL PRODUCT Aldactone® 2. QUALITATIVE AND QUANTITATIVE COMPOSITION Active ingredient: spironolactone 25 mg. 3. PHARMACEUTICAL FORM Tablets are for oral administration. 4. CLINICAL PARTICULARS 4.1 Therapeutic indications Spironolactone is indicated for the following: Congestive cardiac failure. Hepatic cirrhosis with ascites and edema. Malignant ascites. Nephrotic syndrome. Diagnosis and treatment of primary hyperaldosteronism. 4.2 Posology and method of administration Administration of spironolactone once daily with a meal is recommended. Adults Congestive cardiac failure The usual dosage is 100 mg/day. In difficult or severe cases, the dosage may be gradually increased up to 400 mg/day. When edema is controlled, the usual maintenance level is 75 mg/day to 200 mg/day. Hepatic cirrhosis with ascites and edema If urinary Na+/K+ ratio is greater than 1.0, the usual adult dose is 100 mg/day. If the ratio is less than 1.0, the usual adult dose is 200 mg/day to 400 mg/day. Maintenance dosage should be individually determined. Malignant ascites Initial dose is usually 100 mg/day to 200 mg/day. In severe cases the dosage may be gradually increased up to 400 mg/day. When edema is controlled, maintenance dosage should be individually determined. Page 1 of 8 Nephrotic syndrome The usual adult dose is 100 mg/day to 200 mg/day. Spironolactone has not been shown to be anti-inflammatory, or to affect the basic pathological process. Its use is only advised if glucocorticoids by themselves are insufficiently effective. Diagnosis and treatment of primary hyperaldosteronism Spironolactone may be employed as an initial diagnostic measure to provide presumptive evidence of primary hyperaldosteronism while patients are on normal diets. -

Spironolactone
SPIRONOLACTONE This substance was considered by previous working groups, in 1980 (IARC, 1980) and 1987 (IARC, 1987). Since that time, new data have become available, and these have been incorporated into the monograph and taken into consideration in the present evaluation. 1. Exposure Data 1.1 Chemical and physical data 1.1.1 Nomenclature Chem. Abstr. Serv. Reg. No.: 52-01-7 Chem. Abstr. Name: (7α,17α)-7-(Acetylthio)-17-hydroxy-3-oxopregn-4-ene-21- carboxylic acid, γ-lactone IUPAC Systematic Name: 17-Hydroxy-7α-mercapto-3-oxo-17α-pregn-4-ene-21- carboxylic acid, γ-lactone, acetate Synonym: 3-(3-Oxo-7α-acetylthio-17β-hydroxy-4-androsten-17α-yl)propionic acid, γ-lactone 1.1.2 Structural and molecular formulae and relative molecular mass O H3C O CH3 H H H O S C CH3 H O C24H32O4S Relative molecular mass: 416.58 –319– 320 IARC MONOGRAPHS VOLUME 79 1.1.3 Chemical and physical properties of the pure substance (a) Description: Light, cream-coloured to light-tan crystalline powder (Gennaro, 1995) (b) Melting-point: 198–207 °C, with decomposition; occasionally shows prelimi- nary melting at about 135 °C, followed by resolidification (US Pharmacopeial Convention, 1999) (c) Spectroscopy data: Infrared, ultraviolet, nuclear magnetic resonance and mass spectral data have been reported (Sutter & Lau, 1975). (d) Solubility: Practically insoluble in water; soluble in chloroform, ethanol and most organic solvents; slightly soluble in fixed oils (Gennaro, 1995; Budavari, 2000) α 20 ° (e) Optical rotation: [ ]D , –33.5 (chloroform) (Budavari, 1998) 1.1.4 Technical products and impurities Spironolactone is available as 25-, 50- and 100-mg tablets (Gennaro, 1995).