Dissertation S-Nitrosylation Mediates Synaptic Plasticity
Total Page:16
File Type:pdf, Size:1020Kb

Load more
Recommended publications
-
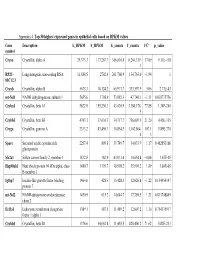
Appendix 4. Top 50 Highest Expressed Genes in Epithelial Cells Based on RPKM Values
Appendix 4. Top 50 highest expressed genes in epithelial cells based on RPKM values Gene Description E_RPKM F_RPKM E_counts F_counts FC* p_value symbol Cryaa Crystallin, alpha A 29,373.3 177,267.7 366,616.4 6,264,319. 17.09 9.11E-118 1 RP23– Long intergenic non-coding RNA 11,888.5 2702.4 261,760.9 134,763.0 −1.94 1 81C12.3 Cryab Crystallin, alpha B 5673.3 10,124.2 65,971.7 333,597.9 5.06 2.71E-43 mt-Nd1 NADH dehydrogenase, subunit 1 5655.6 1798.9 53,082.3 47,748.1 −1.11 0.838775756 Cryba1 Crystallin, beta A1 5622.0 155,230.3 43,420.9 3,380,176. 77.85 1.34E-240 5 Crybb3 Crystallin, beta B3 4743.1 37,636.3 34,717.7 736,007.9 21.20 4.45E-135 Cryga Crystallin, gamma A 2333.2 83,496.3 10,854.5 1,162,864. 107.1 5.89E-270 6 3 Sparc Secreted acidic cysteine rich 2257.4 809.8 39,749.7 34,033.9 −1.17 0.462853166 glycoprotein Slc2a1 Solute carrier family 2, member 1 1832.8 162.9 43,031.4 10,654.8 −4.04 1.67E-05 Hsp90ab1 Heat shock protein 90 kDa alpha, class 1480.7 1139.7 18,998.2 35,901.2 1.89 3.84E-05 B member 1 Igfbp7 Insulin-like growth factor binding 1464.6 428.3 15,428.3 12,626.8 −1.22 0.154954147 protein 7 mt-Nd2 NADH-ubiquinone oxidoreductase 1450.9 615.2 14,644.7 17,789.5 1.21 0.833748849 chain 2 Eef1a1 Eukaryotic translation elongation 1389.1 587.5 11,489.2 12,607.2 1.10 0.754135917 factor 1 alpha 1 Crybb1 Crystallin, beta B1 1376.6 34,662.8 11,455.5 820,406.2 71.62 5.82E-233 Htra3 HtrA serine peptidase 3 1338.6 162.0 23,197.6 6433.9 −3.61 3.93E-05 Gnb2l1 Guanine nucleotide-binding protein 1293.3 670.1 14,495.1 21,652.1 1.49 0.001685952 -

Seq2pathway Vignette
seq2pathway Vignette Bin Wang, Xinan Holly Yang, Arjun Kinstlick May 19, 2021 Contents 1 Abstract 1 2 Package Installation 2 3 runseq2pathway 2 4 Two main functions 3 4.1 seq2gene . .3 4.1.1 seq2gene flowchart . .3 4.1.2 runseq2gene inputs/parameters . .5 4.1.3 runseq2gene outputs . .8 4.2 gene2pathway . 10 4.2.1 gene2pathway flowchart . 11 4.2.2 gene2pathway test inputs/parameters . 11 4.2.3 gene2pathway test outputs . 12 5 Examples 13 5.1 ChIP-seq data analysis . 13 5.1.1 Map ChIP-seq enriched peaks to genes using runseq2gene .................... 13 5.1.2 Discover enriched GO terms using gene2pathway_test with gene scores . 15 5.1.3 Discover enriched GO terms using Fisher's Exact test without gene scores . 17 5.1.4 Add description for genes . 20 5.2 RNA-seq data analysis . 20 6 R environment session 23 1 Abstract Seq2pathway is a novel computational tool to analyze functional gene-sets (including signaling pathways) using variable next-generation sequencing data[1]. Integral to this tool are the \seq2gene" and \gene2pathway" components in series that infer a quantitative pathway-level profile for each sample. The seq2gene function assigns phenotype-associated significance of genomic regions to gene-level scores, where the significance could be p-values of SNPs or point mutations, protein-binding affinity, or transcriptional expression level. The seq2gene function has the feasibility to assign non-exon regions to a range of neighboring genes besides the nearest one, thus facilitating the study of functional non-coding elements[2]. Then the gene2pathway summarizes gene-level measurements to pathway-level scores, comparing the quantity of significance for gene members within a pathway with those outside a pathway. -

Supp Table 1.Pdf
Upregulated genes in Hdac8 null cranial neural crest cells fold change Gene Symbol Gene Title 134.39 Stmn4 stathmin-like 4 46.05 Lhx1 LIM homeobox protein 1 31.45 Lect2 leukocyte cell-derived chemotaxin 2 31.09 Zfp108 zinc finger protein 108 27.74 0710007G10Rik RIKEN cDNA 0710007G10 gene 26.31 1700019O17Rik RIKEN cDNA 1700019O17 gene 25.72 Cyb561 Cytochrome b-561 25.35 Tsc22d1 TSC22 domain family, member 1 25.27 4921513I08Rik RIKEN cDNA 4921513I08 gene 24.58 Ofa oncofetal antigen 24.47 B230112I24Rik RIKEN cDNA B230112I24 gene 23.86 Uty ubiquitously transcribed tetratricopeptide repeat gene, Y chromosome 22.84 D8Ertd268e DNA segment, Chr 8, ERATO Doi 268, expressed 19.78 Dag1 Dystroglycan 1 19.74 Pkn1 protein kinase N1 18.64 Cts8 cathepsin 8 18.23 1500012D20Rik RIKEN cDNA 1500012D20 gene 18.09 Slc43a2 solute carrier family 43, member 2 17.17 Pcm1 Pericentriolar material 1 17.17 Prg2 proteoglycan 2, bone marrow 17.11 LOC671579 hypothetical protein LOC671579 17.11 Slco1a5 solute carrier organic anion transporter family, member 1a5 17.02 Fbxl7 F-box and leucine-rich repeat protein 7 17.02 Kcns2 K+ voltage-gated channel, subfamily S, 2 16.93 AW493845 Expressed sequence AW493845 16.12 1600014K23Rik RIKEN cDNA 1600014K23 gene 15.71 Cst8 cystatin 8 (cystatin-related epididymal spermatogenic) 15.68 4922502D21Rik RIKEN cDNA 4922502D21 gene 15.32 2810011L19Rik RIKEN cDNA 2810011L19 gene 15.08 Btbd9 BTB (POZ) domain containing 9 14.77 Hoxa11os homeo box A11, opposite strand transcript 14.74 Obp1a odorant binding protein Ia 14.72 ORF28 open reading -

A Comprehensive Analysis of the Expression of Crystallins in Mouse Retina Jinghua Xi Washington University School of Medicine in St
Washington University School of Medicine Digital Commons@Becker Open Access Publications 2003 A comprehensive analysis of the expression of crystallins in mouse retina Jinghua Xi Washington University School of Medicine in St. Louis Rafal Farjo University of Michigan - Ann Arbor Shigeo Yoshida University of Michigan - Ann Arbor Timothy S. Kern Case Western Reserve University Anand Swaroop University of Michigan - Ann Arbor See next page for additional authors Follow this and additional works at: https://digitalcommons.wustl.edu/open_access_pubs Recommended Citation Xi, Jinghua; Farjo, Rafal; Yoshida, Shigeo; Kern, Timothy S.; Swaroop, Anand; and Andley, Usha P., ,"A comprehensive analysis of the expression of crystallins in mouse retina." Molecular Vision.9,. 410-419. (2003). https://digitalcommons.wustl.edu/open_access_pubs/1801 This Open Access Publication is brought to you for free and open access by Digital Commons@Becker. It has been accepted for inclusion in Open Access Publications by an authorized administrator of Digital Commons@Becker. For more information, please contact [email protected]. Authors Jinghua Xi, Rafal Farjo, Shigeo Yoshida, Timothy S. Kern, Anand Swaroop, and Usha P. Andley This open access publication is available at Digital Commons@Becker: https://digitalcommons.wustl.edu/open_access_pubs/1801 Molecular Vision 2003; 9:410-9 <http://www.molvis.org/molvis/v9/a53> © 2003 Molecular Vision Received 28 May 2003 | Accepted 19 August 2003 | Published 28 August 2003 A comprehensive analysis of the expression of crystallins in mouse retina Jinghua Xi,1 Rafal Farjo,3 Shigeo Yoshida,3 Timothy S. Kern,5 Anand Swaroop,3,4 Usha P. Andley1,2 Departments of 1Ophthalmology and Visual Sciences and 2Biochemistry and Molecular Biophysics, Washington University School of Medicine, St. -

Genomic Aberrations Associated with Erlotinib Resistance in Non-Small Cell Lung Cancer Cells
ANTICANCER RESEARCH 33: 5223-5234 (2013) Genomic Aberrations Associated with Erlotinib Resistance in Non-small Cell Lung Cancer Cells MASAKUNI SERIZAWA1, TOSHIAKI TAKAHASHI2, NOBUYUKI YAMAMOTO2,3 and YASUHIRO KOH1 1Drug Discovery and Development Division, Shizuoka Cancer Center Research Institute, Sunto-gun, Shizuoka, Japan; 2Division of Thoracic Oncology, Shizuoka Cancer Center Hospital, Sunto-gun, Shizuoka, Japan; 3Third Department of Internal Medicine, Wakayama Medical University, Kimiidera, Wakayama, Japan Abstract. Background/Aim: Mechanisms of resistance to mutations develop resistance, usually within one year of epidermal growth factor receptor (EGFR)-tyrosine kinase treatment. Therefore, there is an urgent need to elucidate the inhibitors (TKIs) in non-small cell lung cancer (NSCLC) underlying mechanisms of resistance in such tumors to are not fully-understood. In this study we aimed to overcome this obstacle (11-14, 17, 24). Recent studies elucidate remaining unknown mechanisms using erlotinib- suggest that mechanisms of acquired resistance to EGFR- resistant NSCLC cells. Materials and Methods: We TKIs can be categorized into three groups: occurrence of performed array comparative genomic hybridization genetic alterations, activation of downstream pathways via (aCGH) to identify genomic aberrations associated with bypass signaling, and phenotypic transformation (15, 16, 21); EGFR-TKI resistance in erlotinib-resistant PC-9ER cells. therapeutic strategies to overcome these resistance Real-time polymerase chain reaction (PCR) and mechanisms are under development. However, although the immunoblot analyses were performed to confirm the results causes of acquired resistance to EGFR-TKIs have been of aCGH. Results: Among the five regions with copy investigated, in more than 30% of patients with acquired number gain detected in PC-9ER cells, we focused on resistance to EGFR-TKI treatment, the mechanisms remain 22q11.2-q12.1 including v-crk avian sarcoma virus CT10 unknown (15). -

Transcriptome Sequencing and Genome-Wide Association Analyses Reveal Lysosomal Function and Actin Cytoskeleton Remodeling in Schizophrenia and Bipolar Disorder
Molecular Psychiatry (2015) 20, 563–572 © 2015 Macmillan Publishers Limited All rights reserved 1359-4184/15 www.nature.com/mp ORIGINAL ARTICLE Transcriptome sequencing and genome-wide association analyses reveal lysosomal function and actin cytoskeleton remodeling in schizophrenia and bipolar disorder Z Zhao1,6,JXu2,6, J Chen3,6, S Kim4, M Reimers3, S-A Bacanu3,HYu1, C Liu5, J Sun1, Q Wang1, P Jia1,FXu2, Y Zhang2, KS Kendler3, Z Peng2 and X Chen3 Schizophrenia (SCZ) and bipolar disorder (BPD) are severe mental disorders with high heritability. Clinicians have long noticed the similarities of clinic symptoms between these disorders. In recent years, accumulating evidence indicates some shared genetic liabilities. However, what is shared remains elusive. In this study, we conducted whole transcriptome analysis of post-mortem brain tissues (cingulate cortex) from SCZ, BPD and control subjects, and identified differentially expressed genes in these disorders. We found 105 and 153 genes differentially expressed in SCZ and BPD, respectively. By comparing the t-test scores, we found that many of the genes differentially expressed in SCZ and BPD are concordant in their expression level (q ⩽ 0.01, 53 genes; q ⩽ 0.05, 213 genes; q ⩽ 0.1, 885 genes). Using genome-wide association data from the Psychiatric Genomics Consortium, we found that these differentially and concordantly expressed genes were enriched in association signals for both SCZ (Po10 − 7) and BPD (P = 0.029). To our knowledge, this is the first time that a substantially large number of genes show concordant expression and association for both SCZ and BPD. Pathway analyses of these genes indicated that they are involved in the lysosome, Fc gamma receptor-mediated phagocytosis, regulation of actin cytoskeleton pathways, along with several cancer pathways. -

AP1B1 Rabbit Polyclonal Antibody – TA323221 | Origene
OriGene Technologies, Inc. 9620 Medical Center Drive, Ste 200 Rockville, MD 20850, US Phone: +1-888-267-4436 [email protected] EU: [email protected] CN: [email protected] Product datasheet for TA323221 AP1B1 Rabbit Polyclonal Antibody Product data: Product Type: Primary Antibodies Applications: IHC Recommended Dilution: ELISA: 1:1000-5000, IHC: 1:25-100 Reactivity: Human, Mouse, Rat Host: Rabbit Isotype: IgG Clonality: Polyclonal Immunogen: Synthetic peptide corresponding to a region derived from 18-30 amino acids of Human Adapter-related protein complex 1 subunit beta-1 Formulation: PBS pH7.3, 0.05% NaN3, 50% glycerol Concentration: lot specific Purification: Antigen affinity purification Conjugation: Unconjugated Storage: Store at -20°C as received. Stability: Stable for 12 months from date of receipt. Gene Name: adaptor related protein complex 1 beta 1 subunit Database Link: NP_001118 Entrez Gene 11764 MouseEntrez Gene 29663 RatEntrez Gene 162 Human Q10567 Background: Adaptor protein complex 1 is found at the cytoplasmic face of coated vesicles located at the Golgi complex; where it mediates both the recruitment of clathrin to the membrane and the recognition of sorting signals within the cytosolic tails of transmembrane receptors. This complex is a heterotetramer composed of two large; one medium; and one small adaptin subunit. The protein encoded by this gene serves as one of the large subunits of this complex and is a member of the adaptin protein family. This gene is a candidate meningioma gene. Alternative splicing results in multiple transcript variants. Synonyms: ADTB1; AP105A; BAM22; CLAPB2 Protein Pathways: Lysosome This product is to be used for laboratory only. -

Supplemental Information
Supplemental information Dissection of the genomic structure of the miR-183/96/182 gene. Previously, we showed that the miR-183/96/182 cluster is an intergenic miRNA cluster, located in a ~60-kb interval between the genes encoding nuclear respiratory factor-1 (Nrf1) and ubiquitin-conjugating enzyme E2H (Ube2h) on mouse chr6qA3.3 (1). To start to uncover the genomic structure of the miR- 183/96/182 gene, we first studied genomic features around miR-183/96/182 in the UCSC genome browser (http://genome.UCSC.edu/), and identified two CpG islands 3.4-6.5 kb 5’ of pre-miR-183, the most 5’ miRNA of the cluster (Fig. 1A; Fig. S1 and Seq. S1). A cDNA clone, AK044220, located at 3.2-4.6 kb 5’ to pre-miR-183, encompasses the second CpG island (Fig. 1A; Fig. S1). We hypothesized that this cDNA clone was derived from 5’ exon(s) of the primary transcript of the miR-183/96/182 gene, as CpG islands are often associated with promoters (2). Supporting this hypothesis, multiple expressed sequences detected by gene-trap clones, including clone D016D06 (3, 4), were co-localized with the cDNA clone AK044220 (Fig. 1A; Fig. S1). Clone D016D06, deposited by the German GeneTrap Consortium (GGTC) (http://tikus.gsf.de) (3, 4), was derived from insertion of a retroviral construct, rFlpROSAβgeo in 129S2 ES cells (Fig. 1A and C). The rFlpROSAβgeo construct carries a promoterless reporter gene, the β−geo cassette - an in-frame fusion of the β-galactosidase and neomycin resistance (Neor) gene (5), with a splicing acceptor (SA) immediately upstream, and a polyA signal downstream of the β−geo cassette (Fig. -

NICU Gene List Generator.Xlsx
Neonatal Crisis Sequencing Panel Gene List Genes: A2ML1 - B3GLCT A2ML1 ADAMTS9 ALG1 ARHGEF15 AAAS ADAMTSL2 ALG11 ARHGEF9 AARS1 ADAR ALG12 ARID1A AARS2 ADARB1 ALG13 ARID1B ABAT ADCY6 ALG14 ARID2 ABCA12 ADD3 ALG2 ARL13B ABCA3 ADGRG1 ALG3 ARL6 ABCA4 ADGRV1 ALG6 ARMC9 ABCB11 ADK ALG8 ARPC1B ABCB4 ADNP ALG9 ARSA ABCC6 ADPRS ALK ARSL ABCC8 ADSL ALMS1 ARX ABCC9 AEBP1 ALOX12B ASAH1 ABCD1 AFF3 ALOXE3 ASCC1 ABCD3 AFF4 ALPK3 ASH1L ABCD4 AFG3L2 ALPL ASL ABHD5 AGA ALS2 ASNS ACAD8 AGK ALX3 ASPA ACAD9 AGL ALX4 ASPM ACADM AGPS AMELX ASS1 ACADS AGRN AMER1 ASXL1 ACADSB AGT AMH ASXL3 ACADVL AGTPBP1 AMHR2 ATAD1 ACAN AGTR1 AMN ATL1 ACAT1 AGXT AMPD2 ATM ACE AHCY AMT ATP1A1 ACO2 AHDC1 ANK1 ATP1A2 ACOX1 AHI1 ANK2 ATP1A3 ACP5 AIFM1 ANKH ATP2A1 ACSF3 AIMP1 ANKLE2 ATP5F1A ACTA1 AIMP2 ANKRD11 ATP5F1D ACTA2 AIRE ANKRD26 ATP5F1E ACTB AKAP9 ANTXR2 ATP6V0A2 ACTC1 AKR1D1 AP1S2 ATP6V1B1 ACTG1 AKT2 AP2S1 ATP7A ACTG2 AKT3 AP3B1 ATP8A2 ACTL6B ALAS2 AP3B2 ATP8B1 ACTN1 ALB AP4B1 ATPAF2 ACTN2 ALDH18A1 AP4M1 ATR ACTN4 ALDH1A3 AP4S1 ATRX ACVR1 ALDH3A2 APC AUH ACVRL1 ALDH4A1 APTX AVPR2 ACY1 ALDH5A1 AR B3GALNT2 ADA ALDH6A1 ARFGEF2 B3GALT6 ADAMTS13 ALDH7A1 ARG1 B3GAT3 ADAMTS2 ALDOB ARHGAP31 B3GLCT Updated: 03/15/2021; v.3.6 1 Neonatal Crisis Sequencing Panel Gene List Genes: B4GALT1 - COL11A2 B4GALT1 C1QBP CD3G CHKB B4GALT7 C3 CD40LG CHMP1A B4GAT1 CA2 CD59 CHRNA1 B9D1 CA5A CD70 CHRNB1 B9D2 CACNA1A CD96 CHRND BAAT CACNA1C CDAN1 CHRNE BBIP1 CACNA1D CDC42 CHRNG BBS1 CACNA1E CDH1 CHST14 BBS10 CACNA1F CDH2 CHST3 BBS12 CACNA1G CDK10 CHUK BBS2 CACNA2D2 CDK13 CILK1 BBS4 CACNB2 CDK5RAP2 -

University of California Santa Cruz Sample
UNIVERSITY OF CALIFORNIA SANTA CRUZ SAMPLE-SPECIFIC CANCER PATHWAY ANALYSIS USING PARADIGM A dissertation submitted in partial satisfaction of the requirements for the degree of DOCTOR OF PHILOSOPHY in BIOMOLECULAR ENGINEERING AND BIOINFORMATICS by Stephen C. Benz June 2012 The Dissertation of Stephen C. Benz is approved: Professor David Haussler, Chair Professor Joshua Stuart Professor Nader Pourmand Dean Tyrus Miller Vice Provost and Dean of Graduate Studies Copyright c by Stephen C. Benz 2012 Table of Contents List of Figures v List of Tables xi Abstract xii Dedication xiv Acknowledgments xv 1 Introduction 1 1.1 Identifying Genomic Alterations . 2 1.2 Pathway Analysis . 5 2 Methods to Integrate Cancer Genomics Data 10 2.1 UCSC Cancer Genomics Browser . 11 2.2 BioIntegrator . 16 3 Pathway Analysis Using PARADIGM 20 3.1 Method . 21 3.2 Comparisons . 26 3.2.1 Distinguishing True Networks From Decoys . 27 3.2.2 Tumor versus Normal - Pathways associated with Ovarian Cancer 29 3.2.3 Differentially Regulated Pathways in ER+ve vs ER-ve breast can- cers . 36 3.2.4 Therapy response prediction using pathways (Platinum Free In- terval in Ovarian Cancer) . 38 3.3 Unsupervised Stratification of Cancer Patients by Pathway Activities . 42 4 SuperPathway - A Global Pathway Model for Cancer 51 4.1 SuperPathway in Ovarian Cancer . 55 4.2 SuperPathway in Breast Cancer . 61 iii 4.2.1 Chin-Naderi Cohort . 61 4.2.2 TCGA Breast Cancer . 63 4.3 Cross-Cancer SuperPathway . 67 5 Pathway Analysis of Drug Effects 74 5.1 SuperPathway on Breast Cell Lines . -

Sporadic Autism Exomes Reveal a Highly Interconnected Protein Network of De Novo Mutations
LETTER doi:10.1038/nature10989 Sporadic autism exomes reveal a highly interconnected protein network of de novo mutations Brian J. O’Roak1,LauraVives1, Santhosh Girirajan1,EmreKarakoc1, Niklas Krumm1,BradleyP.Coe1,RoieLevy1,ArthurKo1,CholiLee1, Joshua D. Smith1, Emily H. Turner1, Ian B. Stanaway1, Benjamin Vernot1, Maika Malig1, Carl Baker1, Beau Reilly2,JoshuaM.Akey1, Elhanan Borenstein1,3,4,MarkJ.Rieder1, Deborah A. Nickerson1, Raphael Bernier2, Jay Shendure1 &EvanE.Eichler1,5 It is well established that autism spectrum disorders (ASD) have a per generation, in close agreement with our previous observations4, strong genetic component; however, for at least 70% of cases, the yet in general, higher than previous studies, indicating increased underlying genetic cause is unknown1. Under the hypothesis that sensitivity (Supplementary Table 2 and Supplementary Table 4)7. de novo mutations underlie a substantial fraction of the risk for We also observed complex classes of de novo mutation including: five developing ASD in families with no previous history of ASD or cases of multiple mutations in close proximity; two events consistent related phenotypes—so-called sporadic or simplex families2,3—we with paternal germline mosaicism (that is, where both siblings con- sequenced all coding regions of the genome (the exome) for tained a de novo event observed in neither parent); and nine events parent–child trios exhibiting sporadic ASD, including 189 new showing a weak minor allele profile consistent with somatic mosaicism trios and 20 that were previously reported4. Additionally, we also (Supplementary Table 3 and Supplementary Figs 2 and 3). sequenced the exomes of 50 unaffected siblings corresponding to Of the severe de novo events, 28% (33 of 120) are predicted to these new (n 5 31) and previously reported trios (n 5 19)4, for a truncate the protein. -

A Peripheral Blood Gene Expression Signature to Diagnose Subclinical Acute Rejection
CLINICAL RESEARCH www.jasn.org A Peripheral Blood Gene Expression Signature to Diagnose Subclinical Acute Rejection Weijia Zhang,1 Zhengzi Yi,1 Karen L. Keung,2 Huimin Shang,3 Chengguo Wei,1 Paolo Cravedi,1 Zeguo Sun,1 Caixia Xi,1 Christopher Woytovich,1 Samira Farouk,1 Weiqing Huang,1 Khadija Banu,1 Lorenzo Gallon,4 Ciara N. Magee,5 Nader Najafian,5 Milagros Samaniego,6 Arjang Djamali ,7 Stephen I. Alexander,2 Ivy A. Rosales,8 Rex Neal Smith,8 Jenny Xiang,3 Evelyne Lerut,9 Dirk Kuypers,10,11 Maarten Naesens ,10,11 Philip J. O’Connell,2 Robert Colvin,8 Madhav C. Menon,1 and Barbara Murphy1 Due to the number of contributing authors, the affiliations are listed at the end of this article. ABSTRACT Background In kidney transplant recipients, surveillance biopsies can reveal, despite stable graft function, histologic features of acute rejection and borderline changes that are associated with undesirable graft outcomes. Noninvasive biomarkers of subclinical acute rejection are needed to avoid the risks and costs associated with repeated biopsies. Methods We examined subclinical histologic and functional changes in kidney transplant recipients from the prospective Genomics of Chronic Allograft Rejection (GoCAR) study who underwent surveillance biopsies over 2 years, identifying those with subclinical or borderline acute cellular rejection (ACR) at 3 months (ACR-3) post-transplant. We performed RNA sequencing on whole blood collected from 88 indi- viduals at the time of 3-month surveillance biopsy to identify transcripts associated with ACR-3, developed a novel sequencing-based targeted expression assay, and validated this gene signature in an independent cohort.