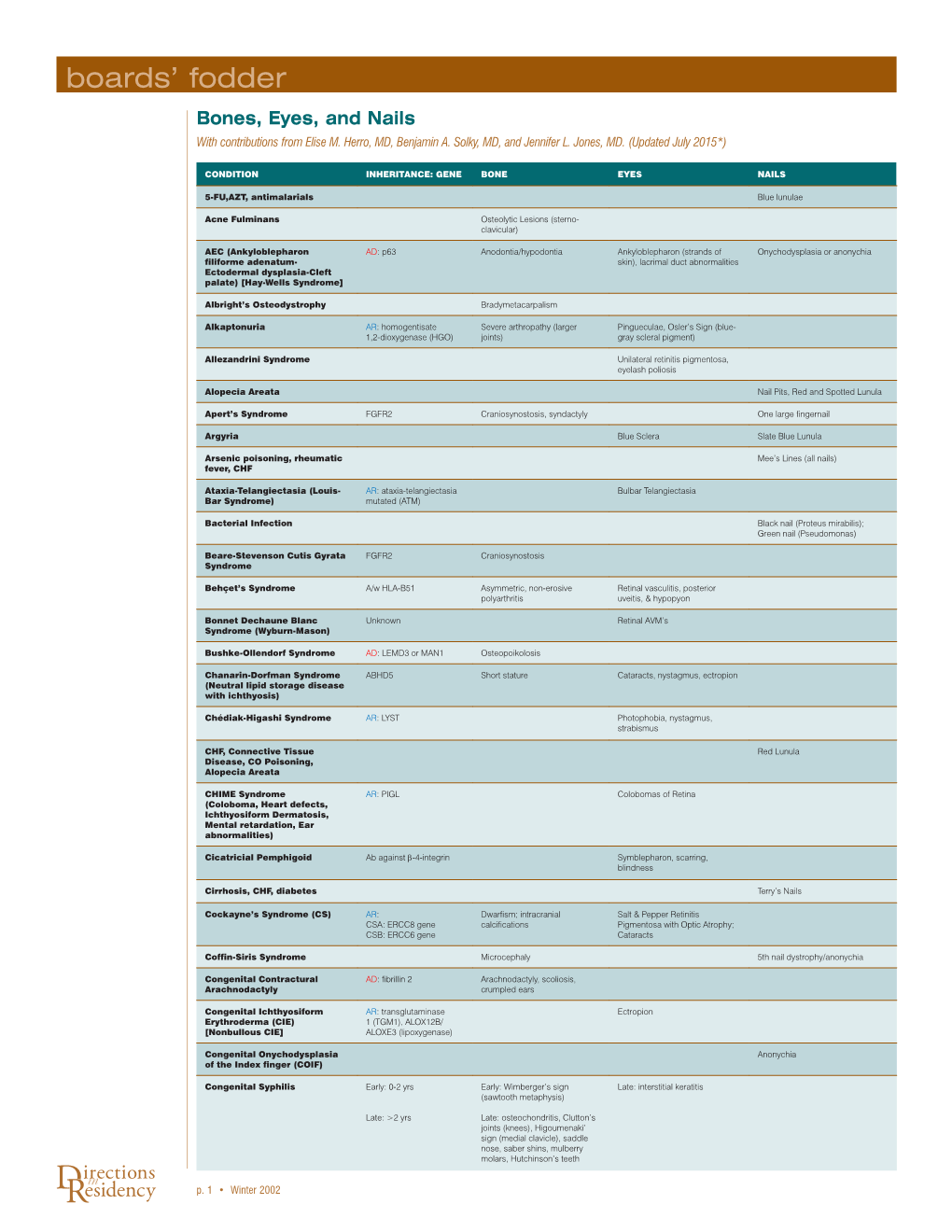
Boards' Fodder
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Genu Varum and Genu Valgum Genu Varum and Genu Valgum
Common Pediatric Lower Limb Disorders Dr.Kholoud Al-Zain Assistant Professor Consultant, Pediatric Orthopedic Surgeon Nov- 2018 Acknowledgement: Dr.Abdalmonem Alsiddiky Dr.Khalid Bakarman Prof. M. Zamzam Topics to Cover 1. In-toeing 2. Genu (varus & valgus), & proximal tibia vara 3. Club foot 4. L.L deformities in C.P patients 5. Limping & leg length inequality 6. Leg aches 1) Intoeing Intoeing- Evaluation • Detailed history – Onset, who noticed it, progression – Fall a lot – How sits on the ground • Screening examination (head to toe) • Pathology at the level of: – Femoral anteversion – Tibial torsion – Forefoot adduction – Wandering big toe Intoeing- Asses rotational profile Pathology Level Special Test • Femoral anteversion • Hips rotational profile: – Supine – Prone • Tibial torsion • Inter-malleolus axis: – Supine – Prone • Foot thigh axis • Forefoot adduction • Heel bisector line • Wandering big toe Intoeing- Special Test Foot Propagation Angle → normal is (-10°) to (+15°) Intoeing- Femoral Anteversion Hips rotational profile, supine → IR/ER normal = 40-45/45-50° Intoeing- Tibial Torsion Inter-malleolus axis Supine position Sitting position Intoeing- Tibial Torsion Foot Thigh Axis → normal (0°) to (-10°) Intoeing- Forefoot Adduction Heel bisector line → normal along 2 toe Intoeing- Adducted Big Toe Intoeing- Treatment • Establish correct diagnosis • Parents education • Annual clinic F/U → asses degree of deformity • Femoral anti-version → sit cross legged • Tibial torsion → spontaneous improvement • Forefoot adduction → anti-version -

Oral Health in Prevalent Types of Ehlers–Danlos Syndromes
View metadata, citation and similar papers at core.ac.uk brought to you by CORE provided by Ghent University Academic Bibliography J Oral Pathol Med (2005) 34: 298–307 ª Blackwell Munksgaard 2005 Æ All rights reserved www.blackwellmunksgaard.com/jopm Oral health in prevalent types of Ehlers–Danlos syndromes Peter J. De Coster1, Luc C. Martens1, Anne De Paepe2 1Department of Paediatric Dentistry, Centre for Special Care, Paecamed Research, Ghent University, Ghent; 2Centre for Medical Genetics, Ghent University Hospital, Ghent, Belgium BACKGROUND: The Ehlers–Danlos syndromes (EDS) Introduction comprise a heterogenous group of heritable disorders of connective tissue, characterized by joint hypermobility, The Ehlers–Danlos syndromes (EDS) comprise a het- skin hyperextensibility and tissue fragility. Most EDS erogenous group of heritable disorders of connective types are caused by mutations in genes encoding different tissue, largely characterized by joint hypermobility, skin types of collagen or enzymes, essential for normal pro- hyperextensibility and tissue fragility (1) (Fig. 1). The cessing of collagen. clinical features, modes of inheritance and molecular METHODS: Oral health was assessed in 31 subjects with bases differ according to the type. EDS are caused by a EDS (16 with hypermobility EDS, nine with classical EDS genetic defect causing an error in the synthesis or and six with vascular EDS), including signs and symptoms processing of collagen types I, III or V. The distribution of temporomandibular disorders (TMD), alterations of and function of these collagen types are displayed in dental hard tissues, oral mucosa and periodontium, and Table 1. At present, two classifications of EDS are was compared with matched controls. -

Viewed Under 23 (B) Or 203 (C) fi M M Male Cko Mice, and Largely Unaffected Magni Cation; Scale Bars, 500 M (B) and 50 M (C)
BRIEF COMMUNICATION www.jasn.org Renal Fanconi Syndrome and Hypophosphatemic Rickets in the Absence of Xenotropic and Polytropic Retroviral Receptor in the Nephron Camille Ansermet,* Matthias B. Moor,* Gabriel Centeno,* Muriel Auberson,* † † ‡ Dorothy Zhang Hu, Roland Baron, Svetlana Nikolaeva,* Barbara Haenzi,* | Natalya Katanaeva,* Ivan Gautschi,* Vladimir Katanaev,*§ Samuel Rotman, Robert Koesters,¶ †† Laurent Schild,* Sylvain Pradervand,** Olivier Bonny,* and Dmitri Firsov* BRIEF COMMUNICATION *Department of Pharmacology and Toxicology and **Genomic Technologies Facility, University of Lausanne, Lausanne, Switzerland; †Department of Oral Medicine, Infection, and Immunity, Harvard School of Dental Medicine, Boston, Massachusetts; ‡Institute of Evolutionary Physiology and Biochemistry, St. Petersburg, Russia; §School of Biomedicine, Far Eastern Federal University, Vladivostok, Russia; |Services of Pathology and ††Nephrology, Department of Medicine, University Hospital of Lausanne, Lausanne, Switzerland; and ¶Université Pierre et Marie Curie, Paris, France ABSTRACT Tight control of extracellular and intracellular inorganic phosphate (Pi) levels is crit- leaves.4 Most recently, Legati et al. have ical to most biochemical and physiologic processes. Urinary Pi is freely filtered at the shown an association between genetic kidney glomerulus and is reabsorbed in the renal tubule by the action of the apical polymorphisms in Xpr1 and primary fa- sodium-dependent phosphate transporters, NaPi-IIa/NaPi-IIc/Pit2. However, the milial brain calcification disorder.5 How- molecular identity of the protein(s) participating in the basolateral Pi efflux remains ever, the role of XPR1 in the maintenance unknown. Evidence has suggested that xenotropic and polytropic retroviral recep- of Pi homeostasis remains unknown. Here, tor 1 (XPR1) might be involved in this process. Here, we show that conditional in- we addressed this issue in mice deficient for activation of Xpr1 in the renal tubule in mice resulted in impaired renal Pi Xpr1 in the nephron. -

Pierre Robin and the Syndrome That Bears His Name PETER RANDALL
Pierre Robin and the Syndrome That Bears His Name PETER RANDALL, M.D. WILTON M. KROGMAN, Ph.D. SOONA JAHINA, B.D.S., M.Sc. Philadelphia, Pennsylvania The Pierre Robin Syndrome refers to a combination of micrognathia (a small jaw) and glossoptosis (literally, a falling downward or back- ward of the tongue) in the newborn infant (Figure 1). These conditions are likely to cause obstruction of the upper airway, and they are fre- quently associated with an incomplete cleft of the palate. Patients with the Pierre Robin Syndrome may present a real emer- gency in the delivery room because of the obstructed upper airway, or the airway problem may not become manifest for several days or weeks (10, 11, 38). There is frequently a feeding problem, as well as problems associated with the cleft of the palate (if one is present) and also an unusual malocclusion (2, 5, 12, 16). In addition, it presents a fascinating anthropological puzzle (22, 23). This paper will review the work of Dr. Robin, consider some possible etiologies of this syndrome, and report on some work on mandibular bone growth in a group of such patients. History Pierre Robin was far from the first person to recognize this syndrome. One account is recorded in 1822 by St. Hilaire. In 1891 Taruffi men- tioned two subclassifications-hypomicrognatus (small jaw) and hypo- agnathus (absent jaw). In 1891, four cases, two of them having cleft palates, were reported by Lanneloague and Monard (12, 14). Shukow- sky in 1902 described a tongue to lip surgical adhesion to overcome the respiratory obstruction (34). -

Tfr2, Hfe, and Hjv in the Regulation of Body Iron Homeostasis
TFR2, HFE, AND HJV IN THE REGULATION OF BODY IRON HOMEOSTASIS By Christal Anna Worthen A DISSERTATION Presented to the Department of Cell & Developmental Biology and the Oregon Health and Science University School of Medicine in partial fulfillment of the requirements for the degree of Doctor of Philosophy June 2014 School of Medicine Oregon Health & Science University CERTIFICATE OF APPROVAL ___________________________________ This is to certify that the PhD dissertation of Christal A Worthen has been approved ______________________________________ Caroline Enns, Ph.D., mentor ______________________________________ Peter Mayinger, Ph.D., Chairman ______________________________________ Philip Stork, M.D. ______________________________________ David Koeller, M.D. ______________________________________ Alex Nechiporuk, Ph.D. TABLE OF CONTENTS i List of Figures ii Acknowledgements iv Abbreviations v Abstract: 1 Chapter 1: Introduction 5 Abstract and Introduction 6 Binding partners, regulation, and trafficking of TFR2 9 Disease-causing mutations in TFR2 12 Hepcidin regulation 12 Physiological function of TFR2 15 Current TFR2 models 18 Summary 19 Figure 21 Chapter 2: The cytoplasmic domain of TFR2 is necessary for 22 HFE, HJV, and TFR2 regulation of hepcidin Abstract 23 Capsule & Introduction 24 Materials and Methods 26 Results 32 Figures 41 Discussion 49 Chapter 3: Lack of functional TFR2 results in stress erythropoiesis 53 Introduction 54 Materials and Methods 55 Results 57 Figures 60 Discussion 65 Chapter 4: Conclusions and future directions 67 Appendices Appendix A: Coculture of HepG2 cells reduces hepcidin expression 71 Appendix B: Hfe-/- macrophages handle iron differently 80 Appendix C: Both ZIP14A and ZIP14B are regulated by HFE and iron 91 Appendix D: The cytoplasmic domain of HFE does not interact with 99 ZIP14 loop 2 by yeast-2-hybris References 105 i LIST OF FIGURES Figure Abstract 1: Body iron homeostasis. -

2021 Follow-Up After Emergency Department Visits for Non-Traumatic Dental Conditions in Adults
DQA Measure EDF-A-A Effective January 1, 2021 **Please read the DQA Measures User Guide prior to implementing this measure.** DQA Measure Specifications: Administrative Claims-Based Measures Follow-up after Emergency Department Visits for Non-Traumatic Dental Conditions in Adults Description: The percentage of ambulatory care sensitive non-traumatic dental condition emergency department visits among adults aged 18 years and older in the reporting period for which the member visited a dentist within (a) 7 days and (b) 30 days of the ED visit Numerators: Number of ambulatory care sensitive non-traumatic dental condition ED visits in the reporting period for which the member visited a dentist within (a) 7 days (NUM1) and (b) 30 days (NUM2) of the ED visit Denominator: Number of ambulatory care sensitive non-traumatic dental condition ED visits in the reporting period Rates: NUM1/DEN and NUM2/DEN Rationale: The use of emergency departments (EDs) for non-traumatic dental conditions has been a growing public health concern across the United States (US)1,2,3,4,5,6,7,8 with over 2 million visits occurring in 2015.9 The majority of ED visits are semi-urgent (53.8%) or non-urgent (23.9%)10, which can be better managed in an ambulatory care setting. Dental care in an ED setting is not definitive with limited care continuity that ultimately leads to poor oral health outcomes.11,12,13 This process of care measure can be used to assess if the patient had timely follow-up with a dentist for more definitive care. References: 1. -

Advances in Understanding the Genetics of Syndromes Involving Congenital Upper Limb Anomalies
Review Article Page 1 of 10 Advances in understanding the genetics of syndromes involving congenital upper limb anomalies Liying Sun1#, Yingzhao Huang2,3,4#, Sen Zhao2,3,4, Wenyao Zhong1, Mao Lin2,3,4, Yang Guo1, Yuehan Yin1, Nan Wu2,3,4, Zhihong Wu2,3,5, Wen Tian1 1Hand Surgery Department, Beijing Jishuitan Hospital, Beijing 100035, China; 2Beijing Key Laboratory for Genetic Research of Skeletal Deformity, Beijing 100730, China; 3Medical Research Center of Orthopedics, Chinese Academy of Medical Sciences, Beijing 100730, China; 4Department of Orthopedic Surgery, 5Department of Central Laboratory, Peking Union Medical College Hospital, Peking Union Medical College and Chinese Academy of Medical Sciences, Beijing 100730, China Contributions: (I) Conception and design: W Tian, N Wu, Z Wu, S Zhong; (II) Administrative support: All authors; (III) Provision of study materials or patients: All authors; (IV) Collection and assembly of data: Y Huang; (V) Data analysis and interpretation: L Sun; (VI) Manuscript writing: All authors; (VII) Final approval of manuscript: All authors. Correspondence to: Wen Tian. Hand Surgery Department, Beijing Jishuitan Hospital, Beijing 100035, China. Email: [email protected]. Abstract: Congenital upper limb anomalies (CULA) are a common birth defect and a significant portion of complicated syndromic anomalies have upper limb involvement. Mostly the mortality of babies with CULA can be attributed to associated anomalies. The cause of the majority of syndromic CULA was unknown until recently. Advances in genetic and genomic technologies have unraveled the genetic basis of many syndromes- associated CULA, while at the same time highlighting the extreme heterogeneity in CULA genetics. Discoveries regarding biological pathways and syndromic CULA provide insights into the limb development and bring a better understanding of the pathogenesis of CULA. -

Download PDF File
Folia Morphol. Vol. 77, No. 2, pp. 386–392 DOI: 10.5603/FM.a2017.0080 O R I G I N A L A R T I C L E Copyright © 2018 Via Medica ISSN 0015–5659 www.fm.viamedica.pl Incidental imaging findings of congenital rib abnormalities: a case series and review of developmental concepts A.M. Aignătoaei1, C.E. Moldoveanu2, I.-D. Căruntu3, S.E. Giuşcă3, S. Partene Vicoleanu3, A.H. Nedelcu3 1Department of Pathology, “Sf. Spiridon” Clinic Hospital, 1, Independenţei Square, Iaşi, Romania 2Department of Radiology, Pneumology Clinic Hospital, 30, dr. Iosif Cihac Street, Iaşi, Romania 3Department of Morphofunctional Sciences, “Grigore T. Popa” University, 16, University Street, Iaşi, Romania [Received: 12 June 2017; Accepted: 24 July 2017] Background: Congenital rib abnormalities are found in approximately 2% of the general population. Usually, they occur in isolation and are rarely symptomatic, but they can also be associated with other malformations. Materials and methods: We reviewed imaging examinations performed over a period of 2 years (2014–2015), enabling us to identify isolated rib abnormalities in 6 adult patients. Results: The case series consisted in 3 cases with bilateral cervical ribs and 1 case each with bifid rib, costal fusion and rib pseudarthrosis. In all patients, the costal anomalies were discovered incidentally. All rib malformations were detected at thoracic radiography, except for the rib pseudarthrosis, which was identified at computed tomography (CT) scan. Differential diagnosis was made between cer- vical ribs and abnormalities of the C7 transverse process and of the first rib, while the other costal malformations were distinguished from tumoural, traumatic or inflammatory lesions of the chest wall, lung and pleura. -

Genes in Eyecare Geneseyedoc 3 W.M
Genes in Eyecare geneseyedoc 3 W.M. Lyle and T.D. Williams 15 Mar 04 This information has been gathered from several sources; however, the principal source is V. A. McKusick’s Mendelian Inheritance in Man on CD-ROM. Baltimore, Johns Hopkins University Press, 1998. Other sources include McKusick’s, Mendelian Inheritance in Man. Catalogs of Human Genes and Genetic Disorders. Baltimore. Johns Hopkins University Press 1998 (12th edition). http://www.ncbi.nlm.nih.gov/Omim See also S.P.Daiger, L.S. Sullivan, and B.J.F. Rossiter Ret Net http://www.sph.uth.tmc.edu/Retnet disease.htm/. Also E.I. Traboulsi’s, Genetic Diseases of the Eye, New York, Oxford University Press, 1998. And Genetics in Primary Eyecare and Clinical Medicine by M.R. Seashore and R.S.Wappner, Appleton and Lange 1996. M. Ridley’s book Genome published in 2000 by Perennial provides additional information. Ridley estimates that we have 60,000 to 80,000 genes. See also R.M. Henig’s book The Monk in the Garden: The Lost and Found Genius of Gregor Mendel, published by Houghton Mifflin in 2001 which tells about the Father of Genetics. The 3rd edition of F. H. Roy’s book Ocular Syndromes and Systemic Diseases published by Lippincott Williams & Wilkins in 2002 facilitates differential diagnosis. Additional information is provided in D. Pavan-Langston’s Manual of Ocular Diagnosis and Therapy (5th edition) published by Lippincott Williams & Wilkins in 2002. M.A. Foote wrote Basic Human Genetics for Medical Writers in the AMWA Journal 2002;17:7-17. A compilation such as this might suggest that one gene = one disease. -

Saethre-Chotzen Syndrome
Saethre-Chotzen syndrome Authors: Professor L. Clauser1 and Doctor M. Galié Creation Date: June 2002 Update: July 2004 Scientific Editor: Professor Raoul CM. Hennekam 1Department of craniomaxillofacial surgery, St. Anna Hospital and University, Corso Giovecca, 203, 44100 Ferrara, Italy. [email protected] Abstract Keywords Disease name and synonyms Excluded diseases Definition Prevalence Management including treatment Etiology Diagnostic methods Genetic counseling Antenatal diagnosis Unresolved questions References Abstract Saethre-Chotzen Syndrome (SCS) is an inherited craniosynostotic condition, with both premature fusion of cranial sutures (craniostenosis) and limb abnormalities. The most common clinical features, present in more than a third of patients, consist of coronal synostosis, brachycephaly, low frontal hairline, facial asymmetry, hypertelorism, broad halluces, and clinodactyly. The estimated birth incidence is 1/25,000 to 1/50,000 but because the phenotype can be very mild, the entity is likely to be underdiagnosed. SCS is inherited as an autosomal dominant trait with a high penetrance and variable expression. The TWIST gene located at chromosome 7p21-p22, is responsible for SCS and encodes a transcription factor regulating head mesenchyme cell development during cranial tube formation. Some patients with an overlapping SCS phenotype have mutations in the FGFR3 (fibroblast growth factor receptor 3) gene; especially the Pro250Arg mutation in FGFR3 (Muenke syndrome) can resemble SCS to a great extent. Significant intrafamilial -

Crouzon Syndrome with Ophthalmological Complications
Journal of Rawalpindi Medical College (JRMC); 2012;16(1):80-81 Case Report Crouzon Syndrome With Ophthalmological Complications Rahela Nasir Paediatric Department, Capital Hospital, CDA Islamabad. Crouzon Syndrome is characterized by premature bilateral optic atrophy. Other facial features included a craniosynostosis. It has an autosomal dominant inheritance prominent nose and deep and narrow palate. No but represents fresh mutation also. Other craniofacial digital abnormalities were seen. No dental aplasia was abnormalities include ocular proptosis caused by shallow present. Systemic examination revealed no orbits with or without divergent strabismus. There may be abnormality. increased intracranial pressure for which surgical morcellation procedures are indicated. A case of He was diagnosed as a case of Crouzon syndrome craniosynostosis is reported which is diagnosed as Crouzon with ocular complications on clinical basis. He was Syndrome with ocular complications on clinical grounds. investigated for microcephaly and suspected Split craniotomy was performed by a neurosurgeon to craniosynostosis. Radiographs of skull showed small relieve raised intracranial pressure and to enhance brain sized skull with early closure of sutures and fontanelle growth. Crouzon Syndrome was originally described in 1912 suggestive of craniosynstosis(Fig 2). A hammered- by Crouzon in a mother and her daughter. It is an autosomal silver (beaten metal/ copper beaten) appearance was dominant inherited disorder but represents fresh mutation also seen due to raised intracranial pressure and also. Crouzon syndrome is characterized by premature compression of the developing brain on the fused craniosynostosis which is quite variable but the coronal suture is nearly always bilaterally involved. Craniofacial bone. CT scan brain with contrast was done which abnormalities include brachycephaly, shallow orbits and confirmed the features suggestive of craniosynostosis maxillary hypoplasia. -

Peds Ortho: What Is Normal, What Is Not, and When to Refer
Peds Ortho: What is normal, what is not, and when to refer Future of Pedatrics June 10, 2015 Matthew E. Oetgen Benjamin D. Martin Division of Orthopaedic Surgery AGENDA • Definitions • Lower Extremity Deformity • Spinal Alignment • Back Pain LOWER EXTREMITY ALIGNMENT DEFINITIONS coxa = hip genu = knee cubitus = elbow pes = foot varus valgus “bow-legged” “knock-knee” apex away from midline apex toward midline normal varus hip (coxa vara) varus humerus valgus ankle valgus hip (coxa valga) Genu varum (bow-legged) Genu valgum (knock knee) bow legs and in toeing often together Normal Limb alignment NORMAL < 2 yo physiologic = reassurance, reevaluate @ 2 yo Bow legged 7° knock knee normal Knock knee physiologic = reassurance, reevaluate in future 4 yo abnormal 10 13 yo abnormal + pain 11 Follow-up is essential! 12 Intoeing 1. Femoral anteversion 2. Tibial torsion 3. Metatarsus adductus MOST LIKELY PHYSIOLOGIC AND WILL RESOLVE! BRACES ARE HISTORY! Femoral Anteversion “W” sitters Internal rotation >> External rotation knee caps point in MOST LIKELY PHYSIOLOGIC AND MAY RESOLVE! Internal Tibial Torsion Thigh foot angle MOST LIKELY PHYSIOLOGIC AND WILL RESOLVE BY SCHOOL AGE Foot is rotated inward Internal Tibial Torsion (Fuchs 1996) Metatarsus Adductus • Flexible = correctible • Observe vs. casting CURVED LATERAL BORDER toes point in NOT TO BE CONFUSED WITH… Clubfoot talipes equinovarus adductus internal varus rotation equinus CAN’T DORSIFLEX cavus Clubfoot START19 CASTING JUST AFTER BIRTH Calcaneovalgus Foot • Intrauterine positioning • Resolve