Sodium Channel Blocker Compositions and the Use
Total Page:16
File Type:pdf, Size:1020Kb

Load more
Recommended publications
-

Granisetron "Vianex"
EU‐RISK MANAGEMENT PLAN GRANISETRON VIANEX® 1 MG/ML, SOLUTION FOR INJECTION/ INFUSION precautionary measure, breast‐feeding should not be advised during treatment with Granisetron “Vianex”. Legal Status: Prescription only product. VI.2 Elements for a public summary VI.2.1 Overview of disease epidemiology Nausea and vomiting associated with chemotherapy and radiotheraphy: One of the most distressing symptoms for patients undergoing both surgery and chemotherapy is nausea and vomiting. These symptoms have a significant impact on quality of life and can lead to malnutrition, inability to respond to treatment and an increased length of hospitalization. Emesis is more commonly associated with chemotherapeutic agents; however, radiation‐induced nausea and vomiting (RINV) can affect a significant proportion of patients, depending on the treated area, dose fractionation, and volume of radiotherapy. The relative risk for developing nausea and vomiting with chemotherapy ranges from 30 to 90% and is dependent upon the chemotherapeutic agent used. Relative risk for nausea and vomiting with radiation therapy is approximately 40%.2,3,4,5 Post‐operative nausea and vomiting Postoperative nausea and vomiting (PONV) is a major source of patient dissatisfaction and is the leading cause of discharge delays and unanticipated postsurgical hospital admissions. In the absence of pharmacological treatment, the rate of PONV is approximately 30% in general population, and can be as high as 70% in patients at highest risk. Several risk factors as surgery type, female gender, non‐smoker status, history of postoperative nausea and vomiting or motion sickness and post‐operative opioid use have been acknowledged. Additionally, post‐ operative vomiting (POV) occurs twice as frequently in children as in adults, increasing until puberty and then decreasing to adult incidence rates. -

Neurontin (Gabapentin)
Texas Prior Authorization Program Clinical Criteria Drug/Drug Class Gabapentin Clinical Criteria Information Included in this Document Neurontin (gabapentin) • Drugs requiring prior authorization: the list of drugs requiring prior authorization for this clinical criteria • Prior authorization criteria logic: a description of how the prior authorization request will be evaluated against the clinical criteria rules • Logic diagram: a visual depiction of the clinical criteria logic • Supporting tables: a collection of information associated with the steps within the criteria (diagnosis codes, procedure codes, and therapy codes); provided when applicable • References: clinical publications and sources relevant to this clinical criteria Note: Click the hyperlink to navigate directly to that section. Gralise (gabapentin Extended Release) • Drugs requiring prior authorization: the list of drugs requiring prior authorization for this clinical criteria • Prior authorization criteria logic: a description of how the prior authorization request will be evaluated against the clinical criteria rules • Logic diagram: a visual depiction of the clinical criteria logic • Supporting tables: a collection of information associated with the steps within the criteria (diagnosis codes, procedure codes, and therapy codes); provided when applicable • References: clinical publications and sources relevant to this clinical criteria Note: Click the hyperlink to navigate directly to that section. March 29, 2019 Copyright © 2019 Health Information Designs, LLC 1 Horizant -

Big Pain Assays Aren't a Big Pain with the Raptor Biphenyl LC Column
Featured Application: 231 Pain Management and Drugs of Abuse Compounds in under 10 Minutes by LC-MS/MS Big Pain Assays Aren’t a Big Pain with the Raptor Biphenyl LC Column • 231 compounds, 40+ isobars, 10 drug classes, 22 ESI- compounds in 10 minutes with 1 column. • A Raptor SPP LC column with time-tested Restek Biphenyl selectivity is the most versatile, multiclass-capable LC column available. • Achieve excellent separation of critical isobars with no tailing peaks. • Run fast and reliable high-throughput LC-MS/MS analyses with increased sensitivity using simple mobile phases. The use of pain management drugs is steadily increasing. As a result, hospital and reference labs are seeing an increase in patient samples that must be screened for a wide variety of pain management drugs to prevent drug abuse and to ensure patient safety and adherence to their medication regimen. Thera- peutic drug monitoring can be challenging due to the low cutoff levels, potential matrix interferences, and isobaric drug compounds. To address these chal- lenges, many drug testing facilities are turning to liquid chromatography coupled with mass spectrometry (LC-MS/MS) for its increased speed, sensitivity, and specificity. As shown in the analysis below, Restek’s Raptor Biphenyl column is ideal for developing successful LC-MS/MS pain medication screening methodologies. With its exceptionally high retention and unique selectivity, 231 multiclass drug compounds and metabolites—including over 40 isobars—can be analyzed in just 10 minutes. In addition, separate panels have been optimized on the Raptor Biphenyl column specifically for opioids, antianxiety drugs, barbiturates, NSAIDs and analgesics, antidepressants, antiepileptics, antipsychotics, hallucinogens, and stimulants for use during confirmation and quantitative analyses. -

Subanesthetic Doses of Ketamine Transiently Decrease Serotonin Transporter Activity: a PET Study in Conscious Monkeys
Neuropsychopharmacology (2013) 38, 2666–2674 & 2013 American College of Neuropsychopharmacology. All rights reserved 0893-133X/13 www.neuropsychopharmacology.org Subanesthetic Doses of Ketamine Transiently Decrease Serotonin Transporter Activity: A PET Study in Conscious Monkeys 1 1 1 1 1 Shigeyuki Yamamoto , Hiroyuki Ohba , Shingo Nishiyama , Norihiro Harada , Takeharu Kakiuchi , 1 ,2 Hideo Tsukada and Edward F Domino* 1 2 Central Research Laboratory, Hamamatsu Photonics KK, Hamakita, Japan; Department of Pharmacology, University of Michigan, Ann Arbor, MI, USA Subanesthetic doses of ketamine, an N-methyl-D-aspartic acid (NMDA) antagonist, have a rapid antidepressant effect which lasts for up to 2 weeks. However, the neurobiological mechanism regarding this effect remains unclear. In the present study, the effects of subanesthetic doses of ketamine on serotonergic systems in conscious monkey brain were investigated. Five young monkeys 11 underwent four positron emission tomography measurements with [ C]-3-amino-4-(2-dimethylaminomethyl-phenylsulfanyl)benzoni- 11 trile ([ C]DASB) for the serotonin transporter (SERT), during and after intravenous infusion of vehicle or ketamine hydrochloride in a 11 dose of 0.5 or 1.5 mg/kg for 40 min, and 24 h post infusion. Global reduction of [ C]DASB binding to SERT was observed during ketamine infusion in a dose-dependent manner, but not 24 h later. The effect of ketamine on the serotonin 1A receptor (5-HT1A-R) and dopamine transporter (DAT) was also investigated in the same subjects studied with [11C]DASB. No significant changes were observed in either 5-HT -R or DAT binding after ketamine infusion. Microdialysis analysis indicated that ketamine infusion transiently increased 1A serotonin levels in the extracellular fluid of the prefrontal cortex. -

SENATE BILL No. 52
As Amended by Senate Committee Session of 2017 SENATE BILL No. 52 By Committee on Public Health and Welfare 1-20 1 AN ACT concerning the uniform controlled substances act; relating to 2 substances included in schedules I, II and V; amending K.S.A. 2016 3 Supp. 65-4105, 65-4107 and 65-4113 and repealing the existing 4 sections. 5 6 Be it enacted by the Legislature of the State of Kansas: 7 Section 1. K.S.A. 2016 Supp. 65-4105 is hereby amended to read as 8 follows: 65-4105. (a) The controlled substances listed in this section are 9 included in schedule I and the number set forth opposite each drug or 10 substance is the DEA controlled substances code which has been assigned 11 to it. 12 (b) Any of the following opiates, including their isomers, esters, 13 ethers, salts, and salts of isomers, esters and ethers, unless specifically 14 excepted, whenever the existence of these isomers, esters, ethers and salts 15 is possible within the specific chemical designation: 16 (1) Acetyl fentanyl (N-(1-phenethylpiperidin-4-yl)- 17 N-phenylacetamide)......................................................................9821 18 (2) Acetyl-alpha-methylfentanyl (N-[1-(1-methyl-2-phenethyl)-4- 19 piperidinyl]-N-phenylacetamide)..................................................9815 20 (3) Acetylmethadol.............................................................................9601 21 (4) AH-7921 (3.4-dichloro-N-[(1- 22 dimethylaminocyclohexylmethyl]benzamide)...............................9551 23 (4)(5) Allylprodine...........................................................................9602 -

THE HARD TRUTH ABOUT PROKINETIC MEDICATION USE in PETS Introduction Pathophysiology/Etiology to That Observed in Dogs
VETTALK Volume 15, Number 04 American College of Veterinary Pharmacists THE HARD TRUTH ABOUT PROKINETIC MEDICATION USE IN PETS Introduction Pathophysiology/Etiology to that observed in dogs. It can be The moving topic of this Vet Talk As with most diseases in the veteri- due to a trichobezoar, dehydration, newsletter will be prokinetic medica- nary world, the etiology and patho- obesity, old age, diabetes, immobility, tions. The availability of information physiology of constipation are varied pain from trauma to the low back, on the many prokinetic agents is var- depending on the species being dis- bladder infection, or an anal sac infec- ied at best so an overall consensus of cussed, where in their gastrointestinal tion. In cases that are more chronic, prokinetic medications will be as- tract the problem is occurring, and underlying disease such as colitis or sessed in this article, hopefully giving any accompanying comorbid condi- Irritable Bowel Syndrome (IBS) may better insight to practitioners about tions. be the culprit. On the other hand, the which agents to use in their patients. cause may be idiopathic which is Canines: In man’s best friend, consti- frustrating for both veterinarian and Prevalence pation has many origins. A dog’s patient since this form is most diffi- Chronic constipation and gastroin- digestive tract itself is complex but cult to treat. testinal stasis are highly debilitating ultimately the mass movements and conditions that not only affect human haustral contractions from the large Equines: Despite their large size, patients but our four legged patients intestine (colon), propel feces into the horses have incredibly delicate diges- as well! Though this condition is rectum stimulating the internal anal tive systems. -

Chapter 25 Mechanisms of Action of Antiepileptic Drugs
Chapter 25 Mechanisms of action of antiepileptic drugs GRAEME J. SILLS Department of Molecular and Clinical Pharmacology, University of Liverpool _________________________________________________________________________ Introduction The serendipitous discovery of the anticonvulsant properties of phenobarbital in 1912 marked the foundation of the modern pharmacotherapy of epilepsy. The subsequent 70 years saw the introduction of phenytoin, ethosuximide, carbamazepine, sodium valproate and a range of benzodiazepines. Collectively, these compounds have come to be regarded as the ‘established’ antiepileptic drugs (AEDs). A concerted period of development of drugs for epilepsy throughout the 1980s and 1990s has resulted (to date) in 16 new agents being licensed as add-on treatment for difficult-to-control adult and/or paediatric epilepsy, with some becoming available as monotherapy for newly diagnosed patients. Together, these have become known as the ‘modern’ AEDs. Throughout this period of unprecedented drug development, there have also been considerable advances in our understanding of how antiepileptic agents exert their effects at the cellular level. AEDs are neither preventive nor curative and are employed solely as a means of controlling symptoms (i.e. suppression of seizures). Recurrent seizure activity is the manifestation of an intermittent and excessive hyperexcitability of the nervous system and, while the pharmacological minutiae of currently marketed AEDs remain to be completely unravelled, these agents essentially redress the balance between neuronal excitation and inhibition. Three major classes of mechanism are recognised: modulation of voltage-gated ion channels; enhancement of gamma-aminobutyric acid (GABA)-mediated inhibitory neurotransmission; and attenuation of glutamate-mediated excitatory neurotransmission. The principal pharmacological targets of currently available AEDs are highlighted in Table 1 and discussed further below. -
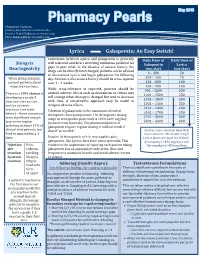
Lyrica Gabapentin: an Easy Switch!
Pharmacist Contacts: [email protected]; [email protected]; [email protected] Lyrica Gabapentin: An Easy Switch! Conversion between Lyrica and gabapentin is generally Daily Dose of Daily Dose of Shingrix well tolerated and direct switching minimizes potential for Gabapentin Lyrica Reactogenicity gaps in pain relief. In the absence of seizure history, the (mg/day) (mg/day) drugs can be directly interchanged; patients can be advised 0 – 300 50 to discontinue Lyrica and begin gabapentin the following When giving Shingrix, day. Patients with a seizure history should be cross-tapered 301 – 450 75 counsel patients about over 1 – 4 weeks. 451 – 600 100 expected reactions. 601 – 900 150 While cross-tolerance is expected, patients should be 901 – 1200 200 advised adverse effects such as drowsiness or edema may There is a 10% chance of 1201 – 1500 250 still emerge when therapy is changed but tend to decrease developing a grade 3 1501 – 1800 300 injection site reaction with time. A conservative approach may be useful to 1801 – 2100 350 and/or systemic mitigate adverse effects. reactions (see table 2101 – 2400 400 Titration of gabapentin to the maximum tolerated 2401 – 2700 450 below) – these symptoms therapeutic dose is important. The therapeutic dosing 2701 – 3000 500 were significant enough range in neuropathic pain trials is 1800-3600 mg/day to prevent regular (normal renal function). The pharmacokinetics of 3001 – 3600 600 activities in about 17% of gabapentin require regular dosing, it will not work if clinical trial patients, but dosed “as needed.” Studies show minimal benefit & tend to pass within 2-3 more adverse effects when high days. -

“GABA” 'Bout? Pregabalin and Gabapentin Abuse
3/19/18 What’s All the “GABA” ‘Bout? Pregabalin and Gabapentin Abuse Courtney Kominek, PharmD, BCPS, CPE Disclosures .Courtney Kominek, PharmD, BCPS, CPE –Axial Healthcare – Consultant .The views and opinions expressed in this presentation are those of the authors and do not necessarily reflect the official policy or position of any agency of the United States government, including the Department of Veterans Affairs. 1 3/19/18 Learning Objectives .Review the proposed mechanisms of action (MOA) for gabapentin and pregabalin. .Explain the proposed rationale as to why gabapentin and pregabalin have become drugs of abuse. .Identify signs and symptoms of withdrawal that an addicted or tolerant patient may experience upon abrupt discontinuation of gabapentin or pregabalin. .Discuss updates on changes in pain management given the increase in gabapentin and pregabalin abuse. Current Situation Opioid overdose public health crisis Rising use of nonopioid medications including gabapentin Opioids and concomitant gabapentin increase risk for overdose Reports of gabapentinoid abuse Changes in PDMP and scheduling at state level http://www.register-herald.com/news/manchin-asks-fda-dea-to-consider-rescheduling-gabapentin/article_442fa04b-7ed9-5bf8-8d19-b5440e9c278b.html 2 3/19/18 Gabapentin and Pregabalin: Pharmacology and Pharmacokinetics Fact or Alternate Fact? .Gabapentin and pregabalin work on GABA. 3 3/19/18 Mechanism of Action Structurally related to GABA and has GABA-mimetic properties Do not • Alter uptake or breakdown • Convert into GABA • Bind to GABAa or GABAB Binds to the α2-δ subunit of the voltage-gated calcium channel Reduces the Ca2+ -dependent release of pro-nociceptive neurotransmitters Decreases release of glutamate, NE, and substance P Dworkin RH et al. -

Comparing Adverse Effects of Analgesic Strategies For
Comparing Adverse Effects daily and weekly time during which higher than that of the only ATC opi- their pain interfered with their mood oids group and 17 times higher than of Analgesic Strategies for or activities, and they indicated the that of the only PRN opioids group. Chronic Cancer Pain amount of relief they received from Source: J Pain Symptom Manage. 2007;33(1):67–77. their pain medicine in the previ- Nausea, vomiting, drowsiness, lack ous week. They also completed the of energy, urinary retention—some- Karnofsky Performance Status (KPS), Clopidogrel: Best Results times the adverse effects of the anal- which measures patients’ ability to per- Pre- or Post-PCI? gesic medication are enough to derail form activities of daily living and their pain management for patients with need for caregiver assistance. When is the best time to give the anti- cancer. It would help to understand There were no significant differ- platelet clopidogrel to patients sched- the relationships between the type of ences between any of the four groups uled for diagnostic coronary angiogra- analgesic prescription and the preva- in terms of pain intensity or amount of phy—before ad hoc coronary stenting lence and severity of adverse effects. time spent in pain. Total pain interfer- or immediately after? Researchers from But not much information is available, ence scores, however, were significantly University of Debrecen, Debrecen, say researchers from the University higher in the ATC plus PRN opioids Hungary and Medical University of of California, San Francisco; the Uni- group than in the no opioids group. Vienna and Wilhelminenhospital, both versity of Nebraska, Omaha; and the The ATC plus PRN opioids group in Vienna, Austria say starting treat- University of Texas Southwestern Med- also had significantly lower functional ment before percutaneous coronary ical Center, Dallas. -

Potential Cannabis Antagonists for Marijuana Intoxication
Central Journal of Pharmacology & Clinical Toxicology Bringing Excellence in Open Access Review Article *Corresponding author Matthew Kagan, M.D., Cedars-Sinai Medical Center, 8730 Alden Drive, Los Angeles, CA 90048, USA, Tel: 310- Potential Cannabis Antagonists 423-3465; Fax: 310.423.8397; Email: Matthew.Kagan@ cshs.org Submitted: 11 October 2018 for Marijuana Intoxication Accepted: 23 October 2018 William W. Ishak, Jonathan Dang, Steven Clevenger, Shaina Published: 25 October 2018 Ganjian, Samantha Cohen, and Matthew Kagan* ISSN: 2333-7079 Cedars-Sinai Medical Center, USA Copyright © 2018 Kagan et al. Abstract OPEN ACCESS Keywords Cannabis use is on the rise leading to the need to address the medical, psychosocial, • Cannabis and economic effects of cannabis intoxication. While effective agents have not yet been • Cannabinoids implemented for the treatment of acute marijuana intoxication, a number of compounds • Antagonist continue to hold promise for treatment of cannabinoid intoxication. Potential therapeutic • Marijuana agents are reviewed with advantages and side effects. Three agents appear to merit • Intoxication further inquiry; most notably Cannabidiol with some evidence of antipsychotic activity • THC and in addition Virodhamine and Tetrahydrocannabivarin with a similar mixed receptor profile. Given the results of this research, continued development of agents acting on cannabinoid receptors with and without peripheral selectivity may lead to an effective treatment for acute cannabinoid intoxication. Much work still remains to develop strategies that will interrupt and reverse the effects of acute marijuana intoxication. ABBREVIATIONS Therapeutic uses of cannabis include chronic pain, loss of appetite, spasticity, and chemotherapy-associated nausea and CBD: Cannabidiol; CBG: Cannabigerol; THCV: vomiting [8]. Recreational cannabis use is on the rise with more Tetrahydrocannabivarin; THC: Tetrahydrocannabinol states approving its use and it is viewed as no different from INTRODUCTION recreational use of alcohol or tobacco [9]. -

Membrane Stabilizer Medications in the Treatment of Chronic Neuropathic Pain: a Comprehensive Review
Current Pain and Headache Reports (2019) 23: 37 https://doi.org/10.1007/s11916-019-0774-0 OTHER PAIN (A KAYE AND N VADIVELU, SECTION EDITORS) Membrane Stabilizer Medications in the Treatment of Chronic Neuropathic Pain: a Comprehensive Review Omar Viswanath1,2,3 & Ivan Urits4 & Mark R. Jones4 & Jacqueline M. Peck5 & Justin Kochanski6 & Morgan Hasegawa6 & Best Anyama7 & Alan D. Kaye7 Published online: 1 May 2019 # Springer Science+Business Media, LLC, part of Springer Nature 2019 Abstract Purpose of Review Neuropathic pain is often debilitating, severely limiting the daily lives of patients who are affected. Typically, neuropathic pain is difficult to manage and, as a result, leads to progression into a chronic condition that is, in many instances, refractory to medical management. Recent Findings Gabapentinoids, belonging to the calcium channel blocking class of drugs, have shown good efficacy in the management of chronic pain and are thus commonly utilized as first-line therapy. Various sodium channel blocking drugs, belonging to the categories of anticonvulsants and local anesthetics, have demonstrated varying degrees of efficacy in the in the treatment of neurogenic pain. Summary Though there is limited medical literature as to efficacy of any one drug, individualized multimodal therapy can provide significant analgesia to patients with chronic neuropathic pain. Keywords Neuropathic pain . Chronic pain . Ion Channel blockers . Anticonvulsants . Membrane stabilizers Introduction Neuropathic pain, which is a result of nervous system injury or lives of patients who are affected. Frequently, it is difficult to dysfunction, is often debilitating, severely limiting the daily manage and as a result leads to the progression of a chronic condition that is, in many instances, refractory to medical This article is part of the Topical Collection on Other Pain management.