Dynein-2 Intermediate Chains Play Crucial but Distinct Roles in Primary
Total Page:16
File Type:pdf, Size:1020Kb

Load more
Recommended publications
-
Shimada 2017.Pdf
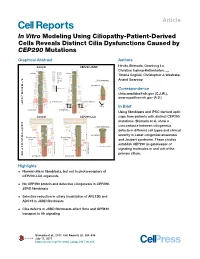
Article In Vitro Modeling Using Ciliopathy-Patient-Derived Cells Reveals Distinct Cilia Dysfunctions Caused by CEP290 Mutations Graphical Abstract Authors Control CEP290-JSRD Hiroko Shimada, Quanlong Lu, Christine Insinna-Kettenhofen, ..., Cilium GPR161 Cilium ADCY3 Tiziana Cogliati, Christopher J. Westlake, Smo ARL13B F CEP290 Cell membrane Anand Swaroop I Gli2 B Cytoplasm R Other proteins O Correspondence Axoneme B Axoneme [email protected] (C.J.W.), L A [email protected] (A.S.) S Cell membrane Cell membrane T Vesicles In Brief Cytoplasm Cytoplasm Using fibroblasts and iPSC-derived optic Control CEP290-LCA cups from patients with distinct CEP290 mutations, Shimada et al. show a P H Cell membrane concordance between ciliogenesis O T ? defects in different cell types and clinical Axoneme ? O ? Axoneme ? Cytoplasm severity in Leber congenital amaurosis R ? Connecting Connecting ? E Cilium Cilium ? and Joubert syndrome. These studies Cell membrane C Cell membrane E establish CEP290 as gatekeeper of P Docked mother centriole T ?? ? ? signaling molecules in and out of the O primary cilium. R Cytoplasm Cytoplasm Highlights d Normal cilia in fibroblasts, but not in photoreceptors of CEP290-LCA organoids d No CEP290 protein and defective ciliogenesis in CEP290- JSRD fibroblasts d Selective reduction in ciliary localization of ARL13B and ADCY3 in JSRD fibroblasts d Cilia defects in JSRD fibroblasts affect Smo and GPR161 transport in Hh signaling Shimada et al., 2017, Cell Reports 20, 384–396 July 11, 2017 http://dx.doi.org/10.1016/j.celrep.2017.06.045 Cell Reports Article In Vitro Modeling Using Ciliopathy-Patient-Derived Cells Reveals Distinct Cilia Dysfunctions Caused by CEP290 Mutations Hiroko Shimada,1 Quanlong Lu,2 Christine Insinna-Kettenhofen,2 Kunio Nagashima,3 Milton A. -

PLATFORM ABSTRACTS Abstract Abstract Numbers Numbers Tuesday, November 6 41
American Society of Human Genetics 62nd Annual Meeting November 6–10, 2012 San Francisco, California PLATFORM ABSTRACTS Abstract Abstract Numbers Numbers Tuesday, November 6 41. Genes Underlying Neurological Disease Room 134 #196–#204 2. 4:30–6:30pm: Plenary Abstract 42. Cancer Genetics III: Common Presentations Hall D #1–#6 Variants Ballroom 104 #205–#213 43. Genetics of Craniofacial and Wednesday, November 7 Musculoskeletal Disorders Room 124 #214–#222 10:30am–12:45 pm: Concurrent Platform Session A (11–19): 44. Tools for Phenotype Analysis Room 132 #223–#231 11. Genetics of Autism Spectrum 45. Therapy of Genetic Disorders Room 130 #232–#240 Disorders Hall D #7–#15 46. Pharmacogenetics: From Discovery 12. New Methods for Big Data Ballroom 103 #16–#24 to Implementation Room 123 #241–#249 13. Cancer Genetics I: Rare Variants Room 135 #25–#33 14. Quantitation and Measurement of Friday, November 9 Regulatory Oversight by the Cell Room 134 #34–#42 8:00am–10:15am: Concurrent Platform Session D (47–55): 15. New Loci for Obesity, Diabetes, and 47. Structural and Regulatory Genomic Related Traits Ballroom 104 #43–#51 Variation Hall D #250–#258 16. Neuromuscular Disease and 48. Neuropsychiatric Disorders Ballroom 103 #259–#267 Deafness Room 124 #52–#60 49. Common Variants, Rare Variants, 17. Chromosomes and Disease Room 132 #61–#69 and Everything in-Between Room 135 #268–#276 18. Prenatal and Perinatal Genetics Room 130 #70–#78 50. Population Genetics Genome-Wide Room 134 #277–#285 19. Vascular and Congenital Heart 51. Endless Forms Most Beautiful: Disease Room 123 #79–#87 Variant Discovery in Genomic Data Ballroom 104 #286–#294 52. -

Unraveling the Genetics of Joubert and Meckel-Gruber Syndromes
Journal of Pediatric Genetics 3 (2014) 65–78 65 DOI 10.3233/PGE-14090 IOS Press Unraveling the genetics of Joubert and Meckel-Gruber syndromes Katarzyna Szymanska, Verity L. Hartill and Colin A. Johnson∗ Department of Ophthalmology and Neuroscience, University of Leeds, Leeds, UK Received 27 May 2014 Revised 11 July 2014 Accepted 14 July 2014 Abstract. Joubert syndrome (JBTS) and Meckel-Gruber syndrome (MKS) are recessive neurodevelopmental conditions caused by mutations in proteins that are structural or functional components of the primary cilium. In this review, we provide an overview of their clinical diagnosis, management and molecular genetics. Both have variable phenotypes, extreme genetic heterogeneity, and display allelism both with each other and other ciliopathies. Recent advances in genetic technology have significantly improved diagnosis and clinical management of ciliopathy patients, with the delineation of some general genotype-phenotype correlations. We highlight those that are most relevant for clinical practice, including the correlation between TMEM67 mutations and the JBTS variant phenotype of COACH syndrome. The subcellular localization of the known MKS and JBTS proteins is now well-described, and we discuss some of the contemporary ideas about ciliopathy disease pathogenesis. Most JBTS and MKS proteins localize to a discrete ciliary compartment called the transition zone, and act as structural components of the so-called “ciliary gate” to regulate the ciliary trafficking of cargo proteins or lipids. Cargo proteins include enzymes and transmembrane proteins that mediate intracellular signaling. The disruption of transition zone function may contribute to the ciliopathy phenotype by altering the composition of the ciliary membrane or axoneme, with impacts on essential developmental signaling including the Wnt and Shh pathways as well as the regulation of secondary messengers such as inositol-1,4,5-trisphosphate (InsP3) and cyclic adenosine monophosphate (cAMP). -

Prenatal Exome Sequencing in Anomalous Fetuses: New Opportunities and Challenges
© American College of Medical Genetics and Genomics ORIGINAL RESEARCH ARTICLE Prenatal exome sequencing in anomalous fetuses: new opportunities and challenges Neeta L. Vora, MD1, Bradford Powell, MD, PhD2, Alicia Brandt, MS2, Natasha Strande, PhD3,4, Emily Hardisty, MS, CGC1, Kelly Gilmore, MS, CGC1, Ann Katherine M. Foreman, MS, CGC2,5, Kirk Wilhelmsen, MD, PhD6, Chris Bizon, PhD6, Jason Reilly, BA6, Phil Owen, BS6, Cynthia M. Powell, MD, MS2,7, Debra Skinner, PhD, MA8, Christine Rini, PhD9, Anne D. Lyerly, MD, MA10, Kim A. Boggess, MD1, Karen Weck, MD3,4, Jonathan S. Berg, MD, PhD2 and James P. Evans, MD, PhD2,10 Purpose: We investigated the diagnostic and clinical performance the following genes: COL1A1, MUSK, KCTD1, RTTN, TMEM67, of exome sequencing in fetuses with sonographic abnormalities PIEZO1 and DYNC2H1. One additional case revealed a de novo with normal karyotype and microarray and, in some cases, normal nonsense mutation in a novel candidate gene (MAP4K4). gene-specific sequencing. The perceived likelihood that exome sequencing would explain Methods: Exome sequencing was performed on DNA from 15 the results (5.2 on a 10-point scale) was higher than the anomalous fetuses and from the peripheral blood of their approximately 30% diagnostic yield discussed in pretest counseling. parents. Parents provided consent to be informed of diagnostic Conclusion: Exome sequencing had diagnostic utility in a highly results in the fetus, medically actionable findings in the parents, select population of fetuses where a genetic diagnosis was highly and their identification as carrier couples for significant auto- suspected. Challenges related to genetics literacy and variant somal recessive conditions. -

Synergistic Genetic Interactions Between Pkhd1 and Pkd1 Result in an ARPKD-Like Phenotype in Murine Models
BASIC RESEARCH www.jasn.org Synergistic Genetic Interactions between Pkhd1 and Pkd1 Result in an ARPKD-Like Phenotype in Murine Models Rory J. Olson,1 Katharina Hopp ,2 Harrison Wells,3 Jessica M. Smith,3 Jessica Furtado,1,4 Megan M. Constans,3 Diana L. Escobar,3 Aron M. Geurts,5 Vicente E. Torres,3 and Peter C. Harris 1,3 Due to the number of contributing authors, the affiliations are listed at the end of this article. ABSTRACT Background Autosomal recessive polycystic kidney disease (ARPKD) and autosomal dominant polycystic kidney disease (ADPKD) are genetically distinct, with ADPKD usually caused by the genes PKD1 or PKD2 (encoding polycystin-1 and polycystin-2, respectively) and ARPKD caused by PKHD1 (encoding fibrocys- tin/polyductin [FPC]). Primary cilia have been considered central to PKD pathogenesis due to protein localization and common cystic phenotypes in syndromic ciliopathies, but their relevance is questioned in the simple PKDs. ARPKD’s mild phenotype in murine models versus in humans has hampered investi- gating its pathogenesis. Methods To study the interaction between Pkhd1 and Pkd1, including dosage effects on the phenotype, we generated digenic mouse and rat models and characterized and compared digenic, monogenic, and wild-type phenotypes. Results The genetic interaction was synergistic in both species, with digenic animals exhibiting pheno- types of rapidly progressive PKD and early lethality resembling classic ARPKD. Genetic interaction be- tween Pkhd1 and Pkd1 depended on dosage in the digenic murine models, with no significant enhancement of the monogenic phenotype until a threshold of reduced expression at the second locus was breached. -

Primary Cilia Control Glucose Homeostasis Via Islet Paracrine Interactions
Primary cilia control glucose homeostasis via islet paracrine interactions Jing W. Hughesa,1, Jung Hoon Choa, Hannah E. Conwaya, Michael R. DiGrucciob, Xue Wen Ngb, Henry F. Rosemana, Damien Abreua, Fumihiko Uranoa, and David W. Pistonb aDepartment of Medicine, Washington University School of Medicine, St. Louis, MO 63110; and bDepartment of Cell Biology and Physiology, Washington University School of Medicine, St. Louis, MO 63110 Edited by C. Ronald Kahn, Harvard Medical School, Boston, MA, and approved March 10, 2020 (received for review January 31, 2020) Pancreatic islets regulate glucose homeostasis through coordinated cellular synchronicity, and both intra- and intercellular signaling actions of hormone-secreting cells. What underlies the function of pathways that govern core islet functions, demonstrating that the islet as a unit is the close approximation and communication primary cilia are required for islet function as a unit and for the among heterogeneous cell populations, but the structural mediators maintenance of energy homeostasis. of islet cellular cross talk remain incompletely characterized. We generated mice specifically lacking β-cell primary cilia, a cellular or- Results ganelle that has been implicated in regulating insulin secretion, and INS1-Cre/IFT88-Flox Mice Lack β-Cell Cilia. To determine the role of found that the β-cell cilia are required for glucose sensing, calcium primary cilia in β-cell function, we generated βCKO mice by influx, insulin secretion, and cross regulation of α-andδ-cells. Pro- crossing INS1-Cre (16) with IFT88-Flox mice (17). The INS1- tein expression profiling in islets confirms perturbation in these Cre strain was chosen based on efficient and selective re- cellular processes and reveals additional targets of cilia-dependent combination in β-cells and established lack of expression in the signaling. -

Ciliopathies Gene Panel
Ciliopathies Gene Panel Contact details Introduction Regional Genetics Service The ciliopathies are a heterogeneous group of conditions with considerable phenotypic overlap. Levels 4-6, Barclay House These inherited diseases are caused by defects in cilia; hair-like projections present on most 37 Queen Square cells, with roles in key human developmental processes via their motility and signalling functions. Ciliopathies are often lethal and multiple organ systems are affected. Ciliopathies are London, WC1N 3BH united in being genetically heterogeneous conditions and the different subtypes can share T +44 (0) 20 7762 6888 many clinical features, predominantly cystic kidney disease, but also retinal, respiratory, F +44 (0) 20 7813 8578 skeletal, hepatic and neurological defects in addition to metabolic defects, laterality defects and polydactyly. Their clinical variability can make ciliopathies hard to recognise, reflecting the ubiquity of cilia. Gene panels currently offer the best solution to tackling analysis of genetically Samples required heterogeneous conditions such as the ciliopathies. Ciliopathies affect approximately 1:2,000 5ml venous blood in plastic EDTA births. bottles (>1ml from neonates) Ciliopathies are generally inherited in an autosomal recessive manner, with some autosomal Prenatal testing must be arranged dominant and X-linked exceptions. in advance, through a Clinical Genetics department if possible. Referrals Amniotic fluid or CV samples Patients presenting with a ciliopathy; due to the phenotypic variability this could be a diverse set should be sent to Cytogenetics for of features. For guidance contact the laboratory or Dr Hannah Mitchison dissecting and culturing, with ([email protected]) / Prof Phil Beales ([email protected]) instructions to forward the sample to the Regional Molecular Genetics Referrals will be accepted from clinical geneticists and consultants in nephrology, metabolic, laboratory for analysis respiratory and retinal diseases. -

Evaluation of Variability in Human Kidney Organoids
ARTICLES https://doi.org/10.1038/s41592-018-0253-2 Evaluation of variability in human kidney organoids Belinda Phipson 1, Pei X. Er1, Alexander N. Combes1,2, Thomas A. Forbes1,3,4, Sara E. Howden1,2, Luke Zappia1,5, Hsan-Jan Yen1, Kynan T. Lawlor1, Lorna J. Hale1,4, Jane Sun6, Ernst Wolvetang6, Minoru Takasato1,7, Alicia Oshlack1,5 and Melissa H. Little 1,2,4* The utility of human pluripotent stem cell–derived kidney organoids relies implicitly on the robustness and transferability of the protocol. Here we analyze the sources of transcriptional variation in a specific kidney organoid protocol. Although individ- ual organoids within a differentiation batch showed strong transcriptional correlation, we noted significant variation between experimental batches, particularly in genes associated with temporal maturation. Single-cell profiling revealed shifts in neph- ron patterning and proportions of component cells. Distinct induced pluripotent stem cell clones showed congruent transcrip- tional programs, with interexperimental and interclonal variation also strongly associated with nephron patterning. Epithelial cells isolated from organoids aligned with total organoids at the same day of differentiation, again implicating relative matura- tion as a confounder. This understanding of experimental variation facilitated an optimized analysis of organoid-based disease modeling, thereby increasing the utility of kidney organoids for personalized medicine and functional genomics. he ability to derive induced pluripotent stem cells (iPSCs) In this study, we provide a comprehensive transcriptional from the somatic cells of patients1, together with directed dif- and morphological evaluation of our kidney organoid protocol. Tferentiation protocols, provides a capacity to model the cell Applying RNA sequencing (RNA-seq) to 57 whole organoids and types affected by disease. -

Cellular Ciliary Phenotyping Indicates Pathogenicity of Novel Variants in IFT140 and Confrms a Mainzer–Saldino Syndrome Diagnosis Machteld M
Oud et al. Cilia (2018) 7:1 https://doi.org/10.1186/s13630-018-0055-2 Cilia SHORT REPORT Open Access Cellular ciliary phenotyping indicates pathogenicity of novel variants in IFT140 and confrms a Mainzer–Saldino syndrome diagnosis Machteld M. Oud1,2* , Brooke L. Latour1,2‡, Zeineb Bakey1,2‡, Stef J. Letteboer1,2, Dorien Lugtenberg1, Ka Man Wu1,2, Elisabeth A. M. Cornelissen3, Helger G. Yntema1,4, Miriam Schmidts1,2,5, Ronald Roepman1,2† and Ernie M. H. F. Bongers1† Abstract Background: Mainzer–Saldino syndrome (MZSDS) is a skeletal ciliopathy and part of the short-rib thoracic dysplasia (SRTD) group of ciliary disorders. The main characteristics of MZSDS are short limbs, mild narrow thorax, blindness, and renal failure. Thus far, variants in two genes are associated with MZSDS: IFT140, and IFT172. In this study, we describe a 1-year-old girl presenting with mild skeletal abnormalities, Leber congenital amaurosis, and bilateral hear- ing difculties. For establishing an accurate diagnosis, we combined clinical, molecular, and functional analyses. Methods: We performed diagnostic whole-exome sequencing (WES) analysis to determine the genetic cause of the disease and analyzed two gene panels, containing all currently known genes in vision disorders, and in hearing impairment. Upon detection of the likely causative variants, ciliary phenotyping was performed in patient urine- derived renal epithelial cells (URECs) and rescue experiments were performed in CRISPR/Cas9-derived Ift140 knock out cells to determine the pathogenicity of the detected variants in vitro. Cilium morphology, cilium length, and intrafa- gellar transport (IFT) were evaluated by immunocytochemistry. Results: Diagnostic WES revealed two novel compound heterozygous variants in IFT140, encoding IFT140. -

Supplementary Materials
Supplementary materials Supplementary Table S1: MGNC compound library Ingredien Molecule Caco- Mol ID MW AlogP OB (%) BBB DL FASA- HL t Name Name 2 shengdi MOL012254 campesterol 400.8 7.63 37.58 1.34 0.98 0.7 0.21 20.2 shengdi MOL000519 coniferin 314.4 3.16 31.11 0.42 -0.2 0.3 0.27 74.6 beta- shengdi MOL000359 414.8 8.08 36.91 1.32 0.99 0.8 0.23 20.2 sitosterol pachymic shengdi MOL000289 528.9 6.54 33.63 0.1 -0.6 0.8 0 9.27 acid Poricoic acid shengdi MOL000291 484.7 5.64 30.52 -0.08 -0.9 0.8 0 8.67 B Chrysanthem shengdi MOL004492 585 8.24 38.72 0.51 -1 0.6 0.3 17.5 axanthin 20- shengdi MOL011455 Hexadecano 418.6 1.91 32.7 -0.24 -0.4 0.7 0.29 104 ylingenol huanglian MOL001454 berberine 336.4 3.45 36.86 1.24 0.57 0.8 0.19 6.57 huanglian MOL013352 Obacunone 454.6 2.68 43.29 0.01 -0.4 0.8 0.31 -13 huanglian MOL002894 berberrubine 322.4 3.2 35.74 1.07 0.17 0.7 0.24 6.46 huanglian MOL002897 epiberberine 336.4 3.45 43.09 1.17 0.4 0.8 0.19 6.1 huanglian MOL002903 (R)-Canadine 339.4 3.4 55.37 1.04 0.57 0.8 0.2 6.41 huanglian MOL002904 Berlambine 351.4 2.49 36.68 0.97 0.17 0.8 0.28 7.33 Corchorosid huanglian MOL002907 404.6 1.34 105 -0.91 -1.3 0.8 0.29 6.68 e A_qt Magnogrand huanglian MOL000622 266.4 1.18 63.71 0.02 -0.2 0.2 0.3 3.17 iolide huanglian MOL000762 Palmidin A 510.5 4.52 35.36 -0.38 -1.5 0.7 0.39 33.2 huanglian MOL000785 palmatine 352.4 3.65 64.6 1.33 0.37 0.7 0.13 2.25 huanglian MOL000098 quercetin 302.3 1.5 46.43 0.05 -0.8 0.3 0.38 14.4 huanglian MOL001458 coptisine 320.3 3.25 30.67 1.21 0.32 0.9 0.26 9.33 huanglian MOL002668 Worenine -

Restoration of RPGR Expression in Vivo Using CRISPR/Cas9 Gene Editing
Gene Therapy https://doi.org/10.1038/s41434-021-00258-6 ARTICLE Restoration of RPGR expression in vivo using CRISPR/Cas9 gene editing 1 1,2,3 4 5 1 5 4 Jessica D. Gumerson ● Amal Alsufyani ● Wenhan Yu ● Jingqi Lei ● Xun Sun ● Lijin Dong ● Zhijian Wu ● Tiansen Li 1 Received: 13 October 2020 / Revised: 29 March 2021 / Accepted: 1 April 2021 This is a U.S. government work and not under copyright protection in the U.S.; foreign copyright protection may apply 2021 Abstract Mutations in the gene for Retinitis Pigmentosa GTPase Regulator (RPGR) cause the X-linked form of inherited retinal degeneration, and the majority are frameshift mutations in a highly repetitive, purine-rich region of RPGR known as the OFR15 exon. Truncation of the reading frame in this terminal exon ablates the functionally important C-terminal domain. We hypothesized that targeted excision in ORF15 by CRISPR/Cas9 and the ensuing repair by non-homologous end joining could restore RPGR reading frame in a portion of mutant photoreceptors thereby correcting gene function in vivo. We tested this hypothesis in the rd9 mouse, a naturally occurring mutant line that carries a frameshift mutation in RPGRORF15, through rd9 1234567890();,: 1234567890();,: a combination of germline and somatic gene therapy approaches. In germline gene-edited mice, probing with RPGR domain-specific antibodies demonstrated expression of full length RPGRORF15 protein. Hallmark features of RPGR mutation-associated early disease phenotypes, such as mislocalization of cone opsins, were no longer present. Subretinal injections of the same guide RNA (sgRNA) carried in AAV sgRNA and SpCas9 expression vectors restored reading frame of RPGRORF15 in a subpopulation of cells with broad distribution throughout the retina, confirming successful correction of the mutation. -

Blueprint Genetics Short Rib Dysplasia / Asphyxiating Thoracic Dysplasia Panel
Short Rib Dysplasia / Asphyxiating Thoracic Dysplasia Panel Test code: MA1101 Is a 18 gene panel that includes assessment of non-coding variants. Is ideal for patients with a clinical suspicion of asphyxiating thoracic dystrophy or short-rib dysplasia with or without polydactyly. The genes on this panel are included in the Comprehensive Growth Disorders / Skeletal Dysplasias and Disorders Panel. About Short Rib Dysplasia / Asphyxiating Thoracic Dysplasia Short-rib dysplasia (SRD) with or without polydactyly refers to a group of autosomal recessive skeletal ciliopathies that are characterized by a constricted thoracic cage, short ribs, shortened tubular bones, and a trident aspect of the acetabular roof. SRD encompasses Ellis-van Creveld syndrome (EVC), Jeune syndrome or asphyxiating thoracic dystrophy (ATD), short rib- polydactyly syndromes (SRPS, Beemer-Langer type, Majewski type, Saldino-Noonan type, Verma-Naumoff type), and Mainzer-Saldino syndrome (MZSDS). Polydactyly is variably present, and there is phenotypic overlap in the various forms of SRDs, which differ by visceral malformation and metaphyseal appearance. Nonskeletal involvement can include cleft lip/palate as well as anomalies of major organs such as the brain, eye, heart, kidneys, liver, pancreas, intestines, and genitalia. Availability 4 weeks Gene Set Description Genes in the Short Rib Dysplasia / Asphyxiating Thoracic Dysplasia Panel and their clinical significance Gene Associated phenotypes Inheritance ClinVar HGMD CSPP1 Jeune asphyxiating thoracic dystrophy, Joubert