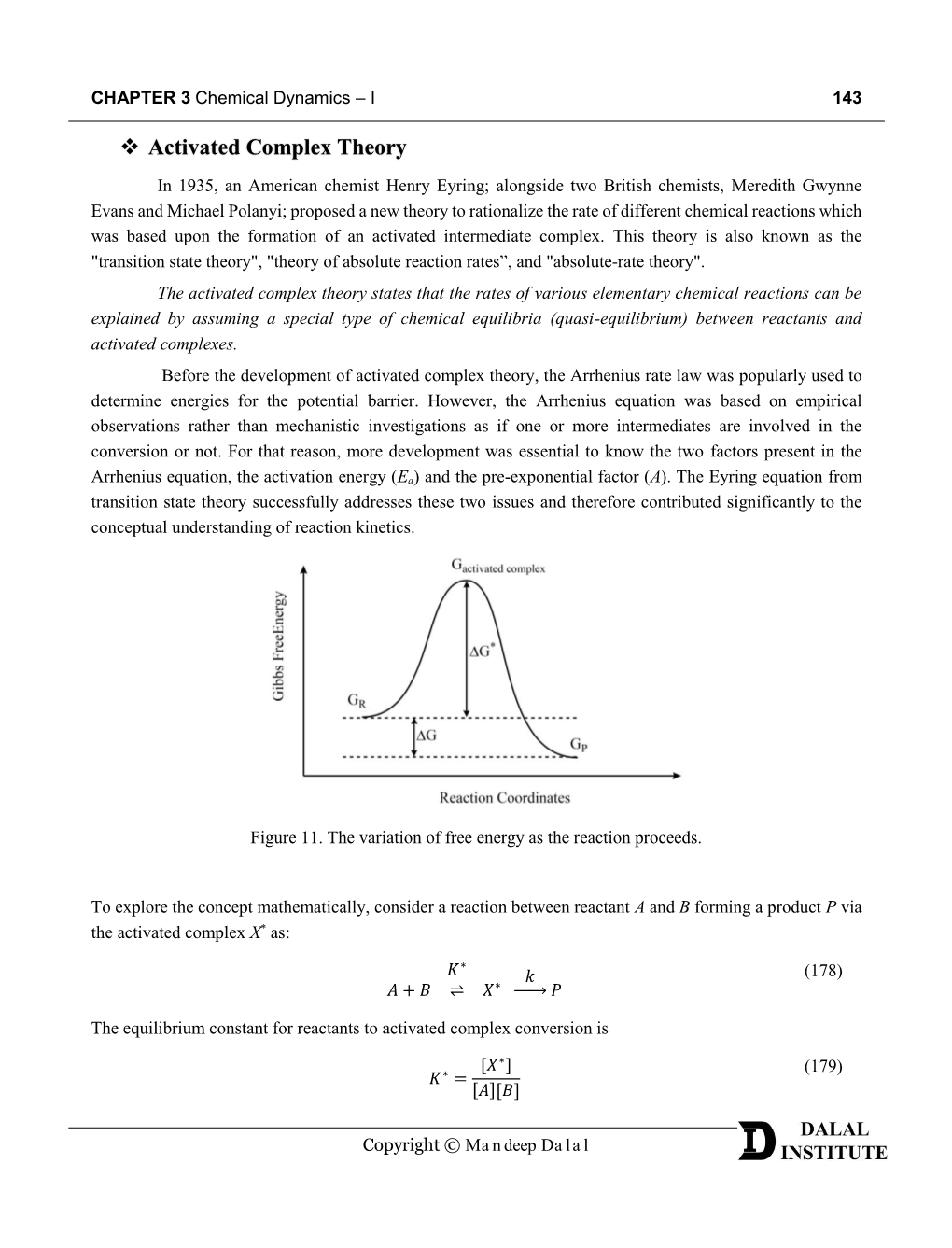
Activated Complex Theory
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Download Author Version (PDF)
Chemical Society Reviews Understanding Catalysis Journal: Chemical Society Reviews Manuscript ID: CS-TRV-06-2014-000210.R2 Article Type: Tutorial Review Date Submitted by the Author: 20-Jun-2014 Complete List of Authors: Roduner, Emil; Universitat Stuttgart, Institut fur Physikalische Chemie Page 1 of 33 Chemical Society Reviews Tutorial Review Understanding Catalysis Emil Roduner Institute of Physical Chemistry, University of Stuttgart, D-70569 Stuttgart, Germany and Chemistry Department, University of Pretoria, Pretoria 0002, Republic of South Africa Abstract The large majority of chemical compounds underwent at least one catalytic step during synthesis. While it is common knowledge that catalysts enhance reaction rates by lowering the activation energy it is often obscure how catalysts achieve this. This tutorial review explains some fundamental principles of catalysis and how the mechanisms are studied. The dissociation of formic acid into H 2 and CO 2, serves to demonstrate how a water molecule can open a new reaction path at lower energy, how immersion in liquid water can influence the charge distribution and energetics, and how catalysis at metal surfaces differs from that in the gas phase. The reversibility of catalytic reactions, the influence of an adsorption pre- equilibrium and the compensating effects of adsorption entropy and enthalpy on the Arrhenius parameters are discussed. It is shown that flexibility around the catalytic centre and residual substrate dynamics on the surface affect these parameters. Sabatier’s principle of optimum substrate adsorption, shape selectivity in the pores of molecular sieves and the polarisation effect at the metal-support interface are explained. Finally, it is shown that the application of a bias voltage in electrochemistry offers an additional parameter to promote or inhibit a reaction. -

Transition State Theory. I
MIT OpenCourseWare http://ocw.mit.edu 5.62 Physical Chemistry II Spring 2008 For information about citing these materials or our Terms of Use, visit: http://ocw.mit.edu/terms. 5.62 Spring 2007 Lecture #33 Page 1 Transition State Theory. I. Transition State Theory = Activated Complex Theory = Absolute Rate Theory ‡ k H2 + F "! [H2F] !!" HF + H Assume equilibrium between reactants H2 + F and the transition state. [H F]‡ K‡ = 2 [H2 ][F] Treat the transition state as a molecule with structure that decays unimolecularly with rate constant k. d[HF] ‡ ‡ = k[H2F] = kK [H2 ][F] dt k has units of sec–1 (unimolecular decay). The motion along the reaction coordinate looks ‡ like an antisymmetric vibration of H2F , one-half cycle of this vibration. Therefore k can be approximated by the frequency of the antisymmetric vibration ν[sec–1] k ≈ ν ≡ frequency of antisymmetric vibration (bond formation and cleavage looks like antisymmetric vibration) d[HF] = νK‡ [H2] [F] dt ! ‡* $ d[HF] (q / N) 'E‡ /kT [ ] = ν # *H F &e [H2 ] F dt "#(q 2 N)(q / N)%& ! ‡ $ ‡ ‡* ‡ (q / N) ' q *' q *' g * ‡ K‡ = # trans &) rot ,) vib ,) 0 ,e-E kT H2 qH2 ) q*H2 , gH2 gF "# (qtrans N) %&( rot +( vib +( 0 0 + ‡ Reaction coordinate is antisymmetric vibrational mode of H2F . This vibration is fully excited (high T limit) because it leads to the cleavage of the H–H bond and the formation of the H–F bond. For a fully excited vibration hν kT The vibrational partition function for the antisymmetric mode is revised 4/24/08 3:50 PM 5.62 Spring 2007 Lecture #33 Page 2 1 kT q*asym = ' since e–hν/kT ≈ 1 – hν/kT vib 1! e!h" kT h! Note that this is an incredibly important simplification. -

Eyring Equation
Search Buy/Sell Used Reactors Glass microreactors Hydrogenation Reactor Buy Or Sell Used Reactors Here. Save Time Microreactors made of glass and lab High performance reactor technology Safe And Money Through IPPE! systems for chemical synthesis scale-up. Worldwide supply www.IPPE.com www.mikroglas.com www.biazzi.com Reactors & Calorimeters Induction Heating Reacting Flow Simulation Steam Calculator For Process R&D Laboratories Check Out Induction Heating Software & Mechanisms for Excel steam table add-in for water Automated & Manual Solutions From A Trusted Source. Chemical, Combustion & Materials and steam properties www.helgroup.com myewoss.biz Processes www.chemgoodies.com www.reactiondesign.com Eyring Equation Peter Keusch, University of Regensburg German version "If the Lord Almighty had consulted me before embarking upon the Creation, I should have recommended something simpler." Alphonso X, the Wise of Spain (1223-1284) "Everything should be made as simple as possible, but not simpler." Albert Einstein Both the Arrhenius and the Eyring equation describe the temperature dependence of reaction rate. Strictly speaking, the Arrhenius equation can be applied only to the kinetics of gas reactions. The Eyring equation is also used in the study of solution reactions and mixed phase reactions - all places where the simple collision model is not very helpful. The Arrhenius equation is founded on the empirical observation that rates of reactions increase with temperature. The Eyring equation is a theoretical construct, based on transition state model. The bimolecular reaction is considered by 'transition state theory'. According to the transition state model, the reactants are getting over into an unsteady intermediate state on the reaction pathway. -

The 'Yard Stick' to Interpret the Entropy of Activation in Chemical Kinetics
World Journal of Chemical Education, 2018, Vol. 6, No. 1, 78-81 Available online at http://pubs.sciepub.com/wjce/6/1/12 ©Science and Education Publishing DOI:10.12691/wjce-6-1-12 The ‘Yard Stick’ to Interpret the Entropy of Activation in Chemical Kinetics: A Physical-Organic Chemistry Exercise R. Sanjeev1, D. A. Padmavathi2, V. Jagannadham2,* 1Department of Chemistry, Geethanjali College of Engineering and Technology, Cheeryal-501301, Telangana, India 2Department of Chemistry, Osmania University, Hyderabad-500007, India *Corresponding author: [email protected] Abstract No physical or physical-organic chemistry laboratory goes without a single instrument. To measure conductance we use conductometer, pH meter for measuring pH, colorimeter for absorbance, viscometer for viscosity, potentiometer for emf, polarimeter for angle of rotation, and several other instruments for different physical properties. But when it comes to the turn of thermodynamic or activation parameters, we don’t have any meters. The only way to evaluate all the thermodynamic or activation parameters is the use of some empirical equations available in many physical chemistry text books. Most often it is very easy to interpret the enthalpy change and free energy change in thermodynamics and the corresponding activation parameters in chemical kinetics. When it comes to interpretation of change of entropy or change of entropy of activation, more often it frightens than enlightens a new teacher while teaching and the students while learning. The classical thermodynamic entropy change is well explained by Atkins [1] in terms of a sneeze in a busy street generates less additional disorder than the same sneeze in a quiet library (Figure 1) [2]. -

Glossary of Terms Used in Photochemistry, 3Rd Edition (IUPAC
Pure Appl. Chem., Vol. 79, No. 3, pp. 293–465, 2007. doi:10.1351/pac200779030293 © 2007 IUPAC INTERNATIONAL UNION OF PURE AND APPLIED CHEMISTRY ORGANIC AND BIOMOLECULAR CHEMISTRY DIVISION* SUBCOMMITTEE ON PHOTOCHEMISTRY GLOSSARY OF TERMS USED IN PHOTOCHEMISTRY 3rd EDITION (IUPAC Recommendations 2006) Prepared for publication by S. E. BRASLAVSKY‡ Max-Planck-Institut für Bioanorganische Chemie, Postfach 10 13 65, 45413 Mülheim an der Ruhr, Germany *Membership of the Organic and Biomolecular Chemistry Division Committee during the preparation of this re- port (2003–2006) was as follows: President: T. T. Tidwell (1998–2003), M. Isobe (2002–2005); Vice President: D. StC. Black (1996–2003), V. T. Ivanov (1996–2005); Secretary: G. M. Blackburn (2002–2005); Past President: T. Norin (1996–2003), T. T. Tidwell (1998–2005) (initial date indicates first time elected as Division member). The list of the other Division members can be found in <http://www.iupac.org/divisions/III/members.html>. Membership of the Subcommittee on Photochemistry (2003–2005) was as follows: S. E. Braslavsky (Germany, Chairperson), A. U. Acuña (Spain), T. D. Z. Atvars (Brazil), C. Bohne (Canada), R. Bonneau (France), A. M. Braun (Germany), A. Chibisov (Russia), K. Ghiggino (Australia), A. Kutateladze (USA), H. Lemmetyinen (Finland), M. Litter (Argentina), H. Miyasaka (Japan), M. Olivucci (Italy), D. Phillips (UK), R. O. Rahn (USA), E. San Román (Argentina), N. Serpone (Canada), M. Terazima (Japan). Contributors to the 3rd edition were: A. U. Acuña, W. Adam, F. Amat, D. Armesto, T. D. Z. Atvars, A. Bard, E. Bill, L. O. Björn, C. Bohne, J. Bolton, R. Bonneau, H. -

Eyring Activation Energy Analysis of Acetic Anhydride Hydrolysis in Acetonitrile Cosolvent Systems Nathan Mitchell East Tennessee State University
East Tennessee State University Digital Commons @ East Tennessee State University Electronic Theses and Dissertations Student Works 5-2018 Eyring Activation Energy Analysis of Acetic Anhydride Hydrolysis in Acetonitrile Cosolvent Systems Nathan Mitchell East Tennessee State University Follow this and additional works at: https://dc.etsu.edu/etd Part of the Analytical Chemistry Commons Recommended Citation Mitchell, Nathan, "Eyring Activation Energy Analysis of Acetic Anhydride Hydrolysis in Acetonitrile Cosolvent Systems" (2018). Electronic Theses and Dissertations. Paper 3430. https://dc.etsu.edu/etd/3430 This Thesis - Open Access is brought to you for free and open access by the Student Works at Digital Commons @ East Tennessee State University. It has been accepted for inclusion in Electronic Theses and Dissertations by an authorized administrator of Digital Commons @ East Tennessee State University. For more information, please contact [email protected]. Eyring Activation Energy Analysis of Acetic Anhydride Hydrolysis in Acetonitrile Cosolvent Systems ________________________ A thesis presented to the faculty of the Department of Chemistry East Tennessee State University In partial fulfillment of the requirements for the degree Master of Science in Chemistry ______________________ by Nathan Mitchell May 2018 _____________________ Dr. Dane Scott, Chair Dr. Greg Bishop Dr. Marina Roginskaya Keywords: Thermodynamic Analysis, Hydrolysis, Linear Solvent Energy Relationships, Cosolvent Systems, Acetonitrile ABSTRACT Eyring Activation Energy Analysis of Acetic Anhydride Hydrolysis in Acetonitrile Cosolvent Systems by Nathan Mitchell Acetic anhydride hydrolysis in water is considered a standard reaction for investigating activation energy parameters using cosolvents. Hydrolysis in water/acetonitrile cosolvent is monitored by measuring pH vs. time at temperatures from 15.0 to 40.0 °C and mole fraction of water from 1 to 0.750. -

Eyring Equation and the Second Order Rate Law: Overcoming a Paradox
Eyring equation and the second order rate law: Overcoming a paradox L. Bonnet∗ CNRS, Institut des Sciences Mol´eculaires, UMR 5255, 33405, Talence, France Univ. Bordeaux, Institut des Sciences Mol´eculaires, UMR 5255, 33405, Talence, France (Dated: March 8, 2016) The standard transition state theory (TST) of bimolecular reactions was recently shown to lead to a rate law in disagreement with the expected second order rate law [Bonnet L., Rayez J.-C. Int. J. Quantum Chem. 2010, 110, 2355]. An alternative derivation was proposed which allows to overcome this paradox. This derivation allows to get at the same time the good rate law and Eyring equation for the rate constant. The purpose of this paper is to provide rigorous and convincing theoretical arguments in order to strengthen these developments and improve their visibility. I. INTRODUCTION A classic result of first year chemistry courses is that for elementary bimolecular gas-phase reactions [1] of the type A + B −→ P roducts, (1) the time-evolution of the concentrations [A] and [B] is given by the second order rate law d[A] − = K[A][B], (2) dt where K is the rate constant. For reactions proceeding through a barrier along the reaction path, Eyring famous equation [2] allows the estimation of K with a considerable success for more than 80 years [3]. This formula is the central result of activated complex theory [2], nowadays called transition state theory (TST) [3–18]. Note that other researchers than Eyring made seminal contributions to the theory at about the same time [3], like Evans and Polanyi [4], Wigner [5, 6], Horiuti [7], etc.. -

Answers to Selected Problems
Answers to Selected Problems Chapter 1 1.2. 0.55 A 1.3. +-y-.z- ++ y+, z+ t-t-y+,~+ -!-!y+.z+ *x+ lx+ t-x+ *s -ls i- s- 1- s+ -l s+ t- s+ BeO triplet CO singlet NO doublet 1.5. "Ionic bonds" using 6p electrons are stronger than those using 6s electrons (Pb2+2Br- p Pb4 +4Br-). "Covalent bonds" with 6s electrons are stronger than those with 6p electrons (PbMe4 > PbMe2)' The energy gap between 6p and 6s orbitals is larger than that for 2p and 2s orbitals. 1.6. SOl = Cda + b + c + d + e + f) S02 = C2(a + b - c - d + e +f) S03 = C3(a - b + c - d + e + f) 541 542 Answers to Selected Problems S04 = C4 (a + b + c + d - e +f) SOs = Cs(a + b - c - d - e +f) SOb = C6 (a - b - c + d - e + f) 1.9. B. c. D d. 0 1.10. There is no vibrational distortion of a homonuclear diatomic that can lower its symmetry. Chapter 2 2.2. Charge density qi = 1; n-bond order Pii = I. 2.3. Il.E = 1l.E,. + IlE,; 1l.E, = 2(1 - i 12) {J. 2.4. Second-order perturbations are not additive. In double union of even AHs, the second order terms are large and cannot be ignored. Chapter 3 3.1. 4 J -jill jm Answers to Selected Problems 543 3.2. a. Ii) I1E n = 2-=( 2 + --3) {J = 0.92{J " ,,/17.7 j17.7 I1Enp = 2 ( --~- + 1 ) {J = 1.28{J j17.7 )17.7 (ii) I1En, = 2 ( -~= + ~-- ) {J = 0.84{J )20.7 fti0 (iii) I1Enp = 2( -- 6- + -~){J = 1.47{J ,,/17.7 v17.7 (iv) I1En, = 2 ( -/ + 2 ) {J = 1.35{J ..;20.7 )20.7 Thus (iii) > (ii) > Ii): (iv) > (ii). -

Bimolecular Reaction Rate Coefficients in the Last Lecture, We Learned Qualitatively the Reaction Mechanisms of Hydrocarbon Combustion
Combustion Chemistry Hai Wang Stanford University 2015 Princeton-CEFRC Summer School On Combustion Course Length: 3 hrs June 22 – 26, 2015 Copyright ©2015 by Hai Wang This material is not to be sold, reproduced or distributed without prior written permission of the owner, Hai Wang. Lecture 4 4. Bimolecular Reaction Rate Coefficients In the last lecture, we learned qualitatively the reaction mechanisms of hydrocarbon combustion. To make this description quantitative, we will need to have a basic knowledge of reaction rate theories. While the rate coefficients of a large number of combustion reactions are experimentally measured, reaction rate theories are often necessary to interpret the experimental data. We shall focus our discussion here to bimolecular reactions of the type A + B → C + D and leave the discussion for unimolecular reactions to a later time. 4.1 Hard Sphere Collision We discussed earlier that an elementary chemical reaction requires molecular collision. The rate coefficient of a bimolecular reaction is essentially the product of reaction probability of two reactants (A and B) upon collision at a given temperature γ(T) and the frequency of collision ZAB, k(T) [A][B] = γ(T) ZAB (4.1) Here we assume that molecules may be described by rigid spheres. Figure 4.1 shows several scenarios of molecular encounter. Starting from the head-on collision, an offset of the two axes of molecular motion leads to off-center collision. Suppose the diameter of A and B are σA and σB, respectively. The limiting off-center collision would have a spacing equal to (σA+σB)/2 between the two axes of molecular motions. -

Dynamic NMR Experiments: -Dr
Determining the Energy of Activation Parameters from Dynamic NMR Experiments: -Dr. Rich Shoemaker (Source: Dynamic NMR Spectroscopy by J. Sandstöm, and me) • The results contained in this document have been published: o Zimmer, Shoemaker, & Ruminski, Inorganica Chimica Acta, 359(2006) 1478-1484 The most common (and oft inaccurate) method of determining activation energy parameters is through the determination (often estimation) of temperature at which the NMR resonances of 2 exchanging species coalesce. The coalescence temperature (Tc) is then used in conjunction with the maximum peak separation in the low-temperature (i.e. slow-exchange) limit (∆ν in Hz). The biggest source of error using the method of coalescence is (1) accurately determining Tc and (2) accurately determining ∆ν. Often, the isotropic chemical shifts of the exchanging species are temperature dependent, so ∆ν changes with temperature. If this happens, then the error in estimation of the activation energy barrier (∆G‡) can be very large. k The rate constant (k ) in these calculations, for nearly all NMR exchange 1 r A B situations, is actually k1+k2 in a system for A exchanging with B, where: k2 The equation to estimate ∆G‡ using the coalescence temperature is: ‡ Tc -3 ∆G = aT 9.972 + log where a = 4.575 x 10 for units of kcal/mol ∆ν a = 1.914 x 10-2 for units of kJ/mol Note: at the coalescence temperature, kc = π∆υ/√2 A second common method of determining the energy of activation (Ea) is by performing an Arrhenius Plot. If one knows the exchange rate constant (kr) at several temperatures (always in Kelvin), one can plot ln(k) vs. -

CHM 8304 Physical Organic Chemistry
CHM 8304 CHM 8304 Physical Organic Chemistry Kinetic analyses Scientific method • proposal of a hypothesis • conduct experiments to test this hypothesis – confirmation – refutation • the trait of refutability is what distinguishes a good scientific hypothesis from a pseudo-hypothesis (see Karl Popper) 2 Kinetic analyses 1 CHM 8304 “Proof” of a mechanism • a mechanism can never really be ‘proven’ – inter alia, it is not directly observable! • one can propose a hypothetical mechanism • one can conduct experiments designed to refute certain hypotheses • one can retain mechanisms that are not refuted • a mechanism can thus become “generally accepted” 3 Mechanistic studies • allow a better comprehension of a reaction, its scope and its utility • require kinetic experiments – rate of disappearance of reactants – rate of appearance of products • kinetic studies are therefore one of the most important disciplines in experimental chemistry 4 Kinetic analyses 2 CHM 8304 Outline: Energy surfaces • based on section 7.1 of A&D – energy surfaces – energy profiles – the nature of a transition state – rates and rate constants – reaction order and rate laws 5 Energy profiles and surfaces • tools for the visualisation of the change of energy as a function of chemical transformations – profiles: • two dimensions • often, energy as a function of ‘reaction coordinate’ – surfaces: • three dimensions • energy as a function of two reaction coordinates 6 Kinetic analyses 3 CHM 8304 Example: SN2 energy profile • one step passing by a single transition state δ- -

Reaction Mechanism of Transition Metal Complexes
CHAPTER 3 Reaction Mechanism of Transition Metal Complexes – I: Inert and Labile Complexes The metal complexes in which the rate of ligand displacement reactions is very fast and hence show high reactivity are called as labile Complexes and this property is termed as lability. On the other hand, the metal complexes in which the rate of ligand displacement reactions is very slow and hence show less reactivity are called as inert complexes and this property is termed as inertness. Thermodynamic stability is the measure of the extent to which the complex will form or will be transformed into another complex when the system has reached equilibrium while kinetic stability refers to the speeds at which these transformations take place. The thermodynamic stability depends upon the energy difference of the reactant and product if the product has less energy than that of the reactant, it will be more stable than the reactant. The thermodynamic stability of metal complexes is calculated by the overall formation constant. If the value of log β is more than 8, the complex is considered as thermodynamically stable. Figure 1. The general reaction coordinates diagram. The kinetic stability of the complex depends upon the activation energy of the reaction. If the activation energy barrier is low, the reaction will take place at a higher speed. These types of complexes are also called as kinetically unstable. If the activation energy barrier is high, the substance will react slowly and will be called as kinetically stabilized or inert. There is no correlation between thermodynamic and kinetic stability. Thermodynamically stable products may labile or inert and the vice versa is also true.