Complement C3-Targeted Gene Therapy Restricts Onset and Progression Of
Total Page:16
File Type:pdf, Size:1020Kb

Load more
Recommended publications
-

Complement Peptide C3a Receptor 1 Promotes Optic Nerve Degeneration in DBA/2J Mice Jeffrey M
Harder et al. Journal of Neuroinflammation (2020) 17:336 https://doi.org/10.1186/s12974-020-02011-z RESEARCH Open Access Complement peptide C3a receptor 1 promotes optic nerve degeneration in DBA/2J mice Jeffrey M. Harder1, Pete A. Williams1,2 , Catherine E. Braine1,3, Hongtian S. Yang1, Jocelyn M. Thomas1, Nicole E. Foxworth1, Simon W. M. John1,4,5* and Gareth R. Howell1,6,7* Abstract Background: The risk of glaucoma increases significantly with age and exposure to elevated intraocular pressure, two factors linked with neuroinflammation. The complement cascade is a complex immune process with many bioactive end-products, including mediators of inflammation. Complement cascade activation has been shown in glaucoma patients and models of glaucoma. However, the function of complement-mediated inflammation in glaucoma is largely untested. Here, the complement peptide C3a receptor 1 was genetically disrupted in DBA/2J mice, an ocular hypertensive model of glaucoma, to test its contribution to neurodegeneration. Methods: A null allele of C3ar1 was backcrossed into DBA/2J mice. Development of iris disease, ocular hypertension, optic nerve degeneration, retinal ganglion cell activity, loss of RGCs, and myeloid cell infiltration in C3ar1-deficient and sufficient DBA/2J mice were compared across multiple ages. RNA sequencing was performed on microglia from primary culture to determine global effects of C3ar1 on microglia gene expression. Results: Deficiency in C3ar1 lowered the risk of degeneration in ocular hypertensive mice without affecting intraocular pressure elevation at 10.5 months of age. Differences were found in the percentage of mice affected, but not in individual characteristics of disease progression. -

WO 2016/147053 Al 22 September 2016 (22.09.2016) P O P C T
(12) INTERNATIONAL APPLICATION PUBLISHED UNDER THE PATENT COOPERATION TREATY (PCT) (19) World Intellectual Property Organization International Bureau (10) International Publication Number (43) International Publication Date WO 2016/147053 Al 22 September 2016 (22.09.2016) P O P C T (51) International Patent Classification: (71) Applicant: RESVERLOGIX CORP. [CA/CA]; 300, A61K 31/551 (2006.01) A61P 37/02 (2006.01) 4820 Richard Road Sw, Calgary, AB, T3E 6L1 (CA). A61K 31/517 (2006.01) C07D 239/91 (2006.01) (72) Inventors: WASIAK, Sylwia; 431 Whispering Water (21) International Application Number: Trail, Calgary, AB, T3Z 3V1 (CA). KULIKOWSKI, PCT/IB20 16/000443 Ewelina, B.; 31100 Swift Creek Terrace, Calgary, AB, T3Z 0B7 (CA). HALLIDAY, Christopher, R.A.; 403 (22) International Filing Date: 138-18th Avenue SE, Calgary, AB, T2G 5P9 (CA). GIL- 10 March 2016 (10.03.2016) HAM, Dean; 249 Scenic View Close NW, Calgary, AB, (25) Filing Language: English T3L 1Y5 (CA). (26) Publication Language: English (81) Designated States (unless otherwise indicated, for every kind of national protection available): AE, AG, AL, AM, (30) Priority Data: AO, AT, AU, AZ, BA, BB, BG, BH, BN, BR, BW, BY, 62/132,572 13 March 2015 (13.03.2015) US BZ, CA, CH, CL, CN, CO, CR, CU, CZ, DE, DK, DM, 62/264,768 8 December 2015 (08. 12.2015) US DO, DZ, EC, EE, EG, ES, FI, GB, GD, GE, GH, GM, GT, [Continued on nextpage] (54) Title: COMPOSITIONS AND THERAPEUTIC METHODS FOR THE TREATMENT OF COMPLEMENT-ASSOCIATED DISEASES (57) Abstract: The invention comprises methods of modulating the complement cascade in a mammal and for treating and/or preventing diseases and disorders as sociated with the complement pathway by administering a compound of Formula I or Formula II, such as, for example, 2-(4-(2-hydroxyethoxy)-3,5-dimethylphenyl)- 5,7-dimethoxyquinazolin-4(3H)-one or a pharmaceutically acceptable salt thereof. -

Active Complement Component 1, Q
APD208Hu01 200µg Active Complement Component 1, Q Subcomponent B (C1qB) Organism Species: Homo sapiens (Human) Instruction manual FOR RESEARCH USE ONLY NOT FOR USE IN CLINICAL DIAGNOSTIC PROCEDURES 1th Edition (Apr, 2016) [ PROPERTIES ] Source: Prokaryotic expression. Host: E. coli Residues: Gln28~Ala253 Tags: Two N-terminal Tags, His-tag and SUMO-tag Purity: >94% Endotoxin Level: <1.0EU per 1μg (determined by the LAL method). Buffer Formulation: 20mM Tris, 150mM NaCl, pH8.0, containing 0.05% sarcosyl and 5% trehalose. Applications: Cell culture; Activity Assays. (May be suitable for use in other assays to be determined by the end user.) Predicted isoelectric point: 8.9 Predicted Molecular Mass: 37.5kDa Accurate Molecular Mass: 38kDa as determined by SDS-PAGE reducing conditions. [ USAGE ] Reconstitute in 20mM Tris, 150mM NaCl (pH8.0) to a concentration of 0.1-1.0 mg/mL. Do not vortex. [ STORAGE AND STABILITY ] Storage: Avoid repeated freeze/thaw cycles. Store at 2-8oC for one month. Aliquot and store at -80oC for 12 months. Stability Test: The thermal stability is described by the loss rate. The loss rate was determined by accelerated thermal degradation test, that is, incubate the protein at 37oC for 48h, and no obvious degradation and precipitation were observed. The loss rate is less than 5% within the expiration date under appropriate storage condition. [ SEQUENCE ] [ ACTIVITY ] The complement component 1q (or simply C1q) is a protein complex involved in the complement system, which is part of the innate immune system. C1q together with C1r and C1s form the C1 complex. Antibodies of the adaptive immune system can bind antigen, forming an antigen-antibody complex. -

High-Throughput Proteomic Profiling of the Fish Liver Following Bacterial
Causey et al. BMC Genomics (2018) 19:719 https://doi.org/10.1186/s12864-018-5092-0 RESEARCH ARTICLE Open Access High-throughput proteomic profiling of the fish liver following bacterial infection Dwight R Causey1, Moritz A N Pohl1, David A Stead2, Samuel A M Martin1, Christopher J Secombes1 and Daniel J Macqueen1* Abstract Background: High-throughput proteomics was used to determine the role of the fish liver in defense responses to bacterial infection. This was done using a rainbow trout (Oncorhynchus mykiss) model following infection with Aeromonas salmonicida, the causative agent of furunculosis. The vertebrate liver has multifaceted functions in innate immunity, metabolism, and growth; we hypothesize this tissue serves a dual role in supporting host defense in parallel to metabolic adjustments that promote effectiveimmunefunction.Whilepaststudieshavereported mRNA responses to A. salmonicida in salmonids, the impact of bacterial infection on the liver proteome remains uncharacterized in fish. Results: Rainbow trout were injected with A. salmonicida or PBS (control) and liver extracted 48 h later for analysis on a hybrid quadrupole-Orbitrap mass spectrometer. A label-free method was used for protein abundance profiling, which revealed a strong innate immune response along with evidence to support parallel rewiring of metabolic and growth systems. 3076 proteins were initially identified against all proteins (n = 71,293 RefSeq proteins) annotated in a single high-quality rainbow trout reference genome, of which 2433 were maintained for analysis post-quality filtering. Among the 2433 proteins, 109 showed significant differential abundance following A. salmonicida challenge, including many upregulated complement system and acute phase response proteins, in addition to molecules with putative functions that may support metabolic re-adjustments. -

Deficiency of Complement Component 5 Ameliorates Glaucoma in DBA/2J Mice
Rowan University Rowan Digital Works Faculty Scholarship for the College of Science & Mathematics College of Science & Mathematics 6-27-2013 Deficiency of complement component 5 ameliorates glaucoma in DBA/2J mice Gareth R. Howell Ileana Soto Reyes Rowan University, [email protected] Margaret Ryan Leah C. Graham Richard S. Smith See next page for additional authors Follow this and additional works at: https://rdw.rowan.edu/csm_facpub Part of the Neuroscience and Neurobiology Commons Recommended Citation Howell, G. R., Soto, I., Ryan, M., Graham, L. C., Smith, R. S., & John, S. W. (2013). Deficiency of complement component 5 ameliorates glaucoma in DBA/2J mice. Journal of Neuroinflammation, 10, 76-2094-10-76. This Article is brought to you for free and open access by the College of Science & Mathematics at Rowan Digital Works. It has been accepted for inclusion in Faculty Scholarship for the College of Science & Mathematics by an authorized administrator of Rowan Digital Works. Authors Gareth R. Howell, Ileana Soto Reyes, Margaret Ryan, Leah C. Graham, Richard S. Smith, and Simon W.M. John This article is available at Rowan Digital Works: https://rdw.rowan.edu/csm_facpub/24 Howell et al. Journal of Neuroinflammation 2013, 10:76 JOURNAL OF http://www.jneuroinflammation.com/content/10/1/76 NEUROINFLAMMATION RESEARCH Open Access Deficiency of complement component 5 ameliorates glaucoma in DBA/2J mice Gareth R Howell1*†, Ileana Soto1†, Margaret Ryan1, Leah C Graham1, Richard S Smith1 and Simon WM John1,2,3* Abstract Background: Glaucoma is an age-related neurodegenerative disorder involving the loss of retinal ganglion cells (RGCs), which results in blindness. -
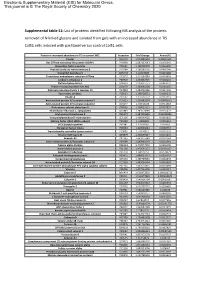
Supplemental Table S1
Electronic Supplementary Material (ESI) for Molecular Omics. This journal is © The Royal Society of Chemistry 2020 Supplemental table S1: List of proteins identified following MS analysis of the proteins removed of N-linked glycans and isolated from gels with an increased abundance in TIS Cal51 cells induced with paclitaxel versus control Cal51 cells. Protein in increased abundance in TIS vs control WCL Accession Fold Change Anova (P) Plectin Q15149 1.073855593 0.00691631 Ras GTPase-activating-like protein IQGAP1 P46940 1.087337643 0.0176342 Elongation factor1-gamma P26641 1.138709703 0.0116496 Peptidyl-prolyl cis-transisomerase B P23284 1.188383105 0.0436246 Dipeptidyl peptidase 3 Q9NY33 1.20163605 0.0215448 Transitional endoplasmic reticulum ATPase P55072 1.214194884 0.0449691 Carbonic anhydrase 2 P00918 1.232852325 0.0158141 Clathrin heavy chain 1 Q00610 1.239621773 0.0463237 Protein transport protein Sec 31A O94979 1.263565104 0.0284155 Aldo-ketoreductase family 1 member C1 Q04828 1.282092186 0.0324406 Spermidine synthase P19623 1.298728621 0.0196232 Plastin-3 P13797 1.310756772 0.0161319 Actin-related protein 2/3 complex subunit 5 O15511 1.333483524 0.00476923 Actin-related protein 2/3 complex subunit 2 O15144 1.35416168 0.0411018 Proteasome subunit alpha type-5 P28066 1.358015551 0.0337657 Thioredoxin reductase 1, cytoplasmic Q16881 1.383670089 0.0235472 Acyl-protein thioesterase 2 O95372 1.387415589 0.00233899 Isoaspartylpeptidase/L-asparaginase Q7L266 1.408149002 0.0319602 Splicing factor U2AF 65kDa subunit P26368 1.41489991 0.0256619 -

In Kidney Injury
antioxidants Review Regulation of Complement Activation by Heme Oxygenase-1 (HO-1) in Kidney Injury Maria G. Detsika 1,* and Elias A. Lianos 2,3 1 First Department of Critical Care Medicine & Pulmonary Services, GP Livanos and M. Simou Laboratories, National & Kapodistrian University of Athens, Medical School, Evangelismos Hospital, 10675 Athens, Greece 2 Thorax Foundation, Research Center of Intensive Care and Emergency Thoracic Medicine, 10675 Athens, Greece; [email protected] 3 Veterans Affairs Medical Center and Virginia Tech, Carilion School of Medicine, 1970 Roanoke Blvd, Salem, VA 24153, USA * Correspondence: [email protected]; Tel.: +30-210-723552; Fax: +30-210-7239127 Abstract: Heme oxygenase is a cytoprotective enzyme with strong antioxidant and anti-apoptotic properties. Its cytoprotective role is mainly attributed to its enzymatic activity, which involves the degradation of heme to biliverdin with simultaneous release of carbon monoxide (CO). Recent studies uncovered a new cytoprotective role for heme oxygenase-1 (HO-1) by identifying a regulatory role on the complement control protein decay-accelerating factor. This is a key complement regulatory protein preventing dysregulation or overactivation of complement cascades that can cause kidney injury. Cell-specific targeting of HO-1 induction may, therefore, be a novel approach to attenuate complement-dependent forms of kidney disease. Keywords: heme; heme oxygenase-1 (HO-1); complement; kidney injury 1. Introduction Citation: Detsika, M.G.; Lianos, E.A. Although the role of heme, in various cellular processes, such as gene transcription Regulation of Complement and translation and cellular differentiation, proliferation, and apoptosis, has been known Activation by Heme Oxygenase-1 for decades, the role of the heme-degrading enzyme heme oxygenase-1 (HO-1) only gained (HO-1) in Kidney Injury. -

Complement Expression in the Retina Is Not Influenced by Short-Term
City University of New York (CUNY) CUNY Academic Works Publications and Research Kingsborough Community College 2014 Complement Expression in the Retina is not Influenced yb Short- term Pressure Elevation Konstantin Astafurov SUNY Downstate Medical Center Cecilia Q. Dong SUNY Downstate Medical Center Lampros Panagis SUNY Downstate Medical Center Gautam Kamtham Mount Sinai School of Medicine Lizhen Ren SUNY Downstate Medical Center See next page for additional authors How does access to this work benefit ou?y Let us know! More information about this work at: https://academicworks.cuny.edu/kb_pubs/46 Discover additional works at: https://academicworks.cuny.edu This work is made publicly available by the City University of New York (CUNY). Contact: [email protected] Authors Konstantin Astafurov, Cecilia Q. Dong, Lampros Panagis, Gautam Kamtham, Lizhen Ren, Anna Rozenboym, Tarique D. Perera, Jeremy D. Coplan, and John Danias This article is available at CUNY Academic Works: https://academicworks.cuny.edu/kb_pubs/46 Molecular Vision 2014; 20:140-152 <http://www.molvis.org/molvis/v20/140> © 2014 Molecular Vision Received 25 February 2013 | Accepted 28 January 2014 | Published 31 January 2014 Complement expression in the retina is not influenced by short- term pressure elevation Konstantin Astafurov,1 Cecilia Q. Dong,2 Lampros Panagis,1 Gautam Kamthan,3 Lizhen Ren,1 Anna Rozenboym,4 Tarique D. Perera,5 Jeremy D. Coplan,6 John Danias1,2 1Department of Cell Biology, SUNY Downstate Medical Center, Brooklyn, NY; 2Department of Ophthalmology, SUNY Downstate Medical Center, Brooklyn, NY; 3Department of Ophthalmology, Mount Sinai School of Medicine, New York, NY; 4Department of Biological Sciences, CUNY Kingsborough Community College, Brooklyn, NY; 5Department of Psychiatry, Columbia University Medical Center and New York State Psychiatric Institute, New York, NY; 6Department of Psychiatry, SUNY Downstate Medical Center, Brooklyn, NY Purpose: To determine whether short-term pressure elevation affects complement gene expression in the retina in vitro and in vivo. -

C1qc) As a Marker for Tuberculosis
Jemds.com Original Research Article Evaluation of Serum Complement Component 1Q Subcomponent C (C1qC) as a Marker for Tuberculosis Ajay Kumar1, Kiran Chawla2, Haritha Madigubba3, Rupesh Thakur4, Dakshina Bisht5 1PhD Scholar, Department of Microbiology, Santosh Medical College and Hospital, Santosh Deemed to be University, NCR Delhi, India. 2Professor & Head, Department of Microbiology, Kasturba Medical College, Manipal Academy of Higher Education, Manipal, Karnataka, India. 3Post Graduate Student, Department of Microbiology, Kasturba Medical College, Manipal Academy of Higher Education, Manipal, Karnataka, India. 4Associate Professor, School of Life & Allied Science, ITM University, Atal Nagar, Raipur, Chattishgarh, India. 5Professor and Head, Department of Microbiology, Santosh Medical College and Hospital, Santosh Deemed to be University, NCR Delhi, India. ABSTRACT BACKGROUND Despite several efforts, tuberculosis (TB) continues to be a global concern. For proper Corresponding Author: management of TB, more sensitive and specific biomarkers for early and accurate Dr. Dakshina Bisht, diagnosis of TB are still required. Blood based host markers are more appealing as an Professor & Head, Department of Microbiology, alternate to sputum-based diagnostics. Thus, in the present study, we evaluated the Santosh Medical College and Hospital, potential of Serum Complement Component 1 Q Subcomponent C (C1qC) as a marker Santosh Deemed to be University, for tuberculosis. NCR Delhi, India. METHODS E-mail: [email protected] Serum samples from 84 subjects which included 39 smear positive pulmonary TB patients and 45 controls (Latent TB n=15, Healthy n= 15 and patients with DOI: 10.14260/jemds/2019/571 respiratory diseases other than TB n= 15) were collected and Enzyme Linked Financial or Other Competing Interests: Immunosorbent Assay was performed to estimate the serum C1qC levels. -

Age-Related Changes in the Retina and the Risk Factors Leading to the Onset of Disease
Age-related changes in the retina and the risk factors leading to the onset of disease Marie Jaimie Hoh Kam A thesis submitted for the degree of Doctor of Philosophy 2011 Supervisor: Professor Glen Jeffery Institute of Ophthalmology University College London Age-related changes in the retina and the risk factors leading to the onset of disease Abstract Age-related macular degeneration (AMD) is the most prevalent form of irreversible blindness in those over 50 years old in Western countries. It is a late- onset, neurodegenerative retinal disease, which is characterised by extracellular deposits containing amyloid beta peptides (Aβ) on the Bruch’s membrane. In half of AMD cases, polymorphisms in the gene encoding complement factor H (CFH) are associated with susceptibility to the disease. The aims of this thesis were; (1) to identify sites of Aβ accumulation in mouse ageing eye and macrophage up-regulation, (2) to investigate the effects of immunotherapy targeting Aβ as a potential treatment for AMD, (3) to examine how pathogens trigger retinal disease in CFH mice, (4) to determine whether the strategy of inhibiting complement component C3 (C3) and complement activation is beneficial or detrimental in CFH mice. I show that Aβ deposition increases with age and is accumulated on photoreceptor outer segment and on Bruch’s membrane. Systemic administration of an antibody targeting Aβ improved retinal pathology, by decreasing deposits and reducing the activation of C3. I also show that genetic mutation or polymorphism is not the only factor triggering the onset of AMD but also environmental factors such as pathogen load are also critical. -

Deciphering Binding Patterns of Therapeutic Antibodies with Immune Cells
Digital Comprehensive Summaries of Uppsala Dissertations from the Faculty of Medicine 1648 Deciphering Binding Patterns of Therapeutic Antibodies with Immune Cells From Method Development to Application SINA BONDZA ACTA UNIVERSITATIS UPSALIENSIS ISSN 1651-6206 ISBN 978-91-513-0902-6 UPPSALA urn:nbn:se:uu:diva-406875 2020 Dissertation presented at Uppsala University to be publicly examined in Rudbecksalen, Rudbecklaboratoriet, Dag Hammarskjölds Väg 20, Uppsala, Thursday, 7 May 2020 at 09:00 for the degree of Doctor of Philosophy (Faculty of Medicine). The examination will be conducted in English. Faculty examiner: Professor Mark Cragg (Academic Unit of Cancer Sciences, University of Southampton). Abstract Bondza, S. 2020. Deciphering Binding Patterns of Therapeutic Antibodies with Immune Cells. From Method Development to Application. Digital Comprehensive Summaries of Uppsala Dissertations from the Faculty of Medicine 1648. 68 pp. Uppsala: Acta Universitatis Upsaliensis. ISBN 978-91-513-0902-6. Reversible binding, for example between signaling molecules and receptors on the cell surface, is one of the main means to communicate information in cellular systems. Knowledge about how molecules interact is crucial for both understanding biological function and for therapeutic intervention. The cellular environment often makes ligand-receptor interactions complex with the membrane providing structural support and containing other components that interfere with the interaction. One of the fastest growing drug classes for targeting cellular receptors are monoclonal antibodies (mAb), in particular within oncology. Therapeutic mAbs can have direct effects on target cells mediated via the Fab-domain and immune-related effects that are mediated via the Fc-domain. An example of the latter is activation of the complement system by binding of its first component C1q to Fc-domains. -

Subretinal Macrophages Produce Classical Complement Activator C1q
Jiao et al. Molecular Neurodegeneration (2018) 13:45 https://doi.org/10.1186/s13024-018-0278-0 RESEARCH ARTICLE Open Access Subretinal macrophages produce classical complement activator C1q leading to the progression of focal retinal degeneration Haihan Jiao1, Matt Rutar1,2, Nilisha Fernando1, Ted Yednock3, Sethu Sankaranarayanan3, Riemke Aggio-Bruce1, Jan Provis1,4 and Riccardo Natoli1,4* Abstract Background: The role of the alternative complement pathway and its mediation by retinal microglia and macrophages, is well-established in the pathogenesis of Age-Related Macular Degeneration (AMD). However, the contribution of the classical complement pathway towards the progression of retinal degenerations is not fully understood, including the role of complement component 1q (C1q) as a critical activator molecule of the classical pathway. Here, we investigated the contribution of C1q to progressive photoreceptor loss and neuroinflammation in retinal degenerations. Methods: Wild-type (WT), C1qa knockout (C1qa−/−) and mice treated with a C1q inhibitor (ANX-M1; Annexon Biosciences), were exposed to photo-oxidative damage (PD) and were observed for progressive lesion development. Retinal function was assessed by electroretinography, followed by histological analyses to assess photoreceptor degeneration. Retinal inflammation was investigated through complement activation, macrophage recruitment and inflammasome expression using western blotting, qPCR and immunofluorescence. C1q was localised in human AMD donor retinas using immunohistochemistry. Results: PD mice had increased levels of C1qa which correlated with increasing photoreceptor cell death and macrophage recruitment. C1qa−/− mice did not show any differences in photoreceptor loss or inflammation at 7 days compared to WT, however at 14 days after the onset of damage, C1qa−/− retinas displayed less photoreceptor cell death, reduced microglia/macrophage recruitment to the photoreceptor lesion, and higher visual function.