Establishing an Automated Graphical Genome Analysis Platform
Total Page:16
File Type:pdf, Size:1020Kb

Load more
Recommended publications
-

A Decision Support System for Reporting High Throughput Sequencing of Cancers in Clinical Diagnostic Laboratories Kenneth D
Doig et al. Genome Medicine (2017) 9:38 DOI 10.1186/s13073-017-0427-z SOFTWARE Open Access PathOS: a decision support system for reporting high throughput sequencing of cancers in clinical diagnostic laboratories Kenneth D. Doig1,2,3,8*, Andrew Fellowes2, Anthony H. Bell2, Andrei Seleznev2, David Ma2, Jason Ellul1, Jason Li1, Maria A. Doyle1, Ella R. Thompson2,3, Amit Kumar1,6,7, Luis Lara1,3, Ravikiran Vedururu2, Gareth Reid2, Thomas Conway1, Anthony T. Papenfuss1,3,5,6 and Stephen B. Fox2,3,4 Abstract Background: The increasing affordability of DNA sequencing has allowed it to be widely deployed in pathology laboratories. However, this has exposed many issues with the analysis and reporting of variants for clinical diagnostic use. Implementing a high-throughput sequencing (NGS) clinical reporting system requires a diverse combination of capabilities, statistical methods to identify variants, global variant databases, a validated bioinformatics pipeline, an auditable laboratory workflow, reproducible clinical assays and quality control monitoring throughout. These capabilities must be packaged in software that integrates the disparate components into a useable system. Results: To meet these needs, we developed a web-based application, PathOS, which takes variant data from a patient sample through to a clinical report. PathOS has been used operationally in the Peter MacCallum Cancer Centre for two years for the analysis, curation and reporting of genetic tests for cancer patients, as well as the curation of large-scale research studies. PathOS has also been deployed in cloud environments allowing multiple institutions to use separate, secure and customisable instances of the system. Increasingly, the bottleneck of variant curation is limiting the adoption of clinical sequencing for molecular diagnostics. -

Locus Reference Genomic Sequences: an Improved Basis for Describing Human DNA Variants
Dalgleish et al. Genome Medicine 2010, 2:24 http://genomemedicine.com/content/2/4/24 CORRESPONDENCE Open Access Locus Reference Genomic sequences: an improved basis for describing human DNA variants Raymond Dalgleish1*, Paul Flicek 2, Fiona Cunningham2, Alex Astashyn3, Raymond E Tully3, Glenn Proctor2, Yuan Chen2, William M McLaren2, Pontus Larsson2, Brendan W Vaughan2, Christophe Béroud4, Glen Dobson5, Heikki Lehväslaiho6, Peter EM Taschner7, Johan T den Dunnen7, Andrew Devereau5, Ewan Birney2, Anthony J Brookes1 and Donna R Maglott3 Introduction Abstract In 1993 Ernest Beutler wrote an eloquent letter to the As our knowledge of the complexity of gene editor of the American Journal of Human Genetics high- architecture grows, and we increase our understanding lighting the defi ciencies of the systems then used to of the subtleties of gene expression, the process of describe DNA variants [1]. Th at same year, the editor of accurately describing disease-causing gene variants Human Mutation invited Arthur Beaudet and Lap-Chee has become increasingly problematic. In part, this is Tsui to produce a nomenclature for variants in genes and due to current reference DNA sequence formats that proteins [2]. From these simple beginnings, the last 17 do not fully meet present needs. Here we present the years have borne witness to the steady development of Locus Reference Genomic (LRG) sequence format, the nomenclature used to describe sequence variation which has been designed for the specifi c purpose that is now maintained under the auspices of the Human of gene variant reporting. The format builds on Genome Variation Society (HGVS) [3,4]. To some, the the successful National Center for Biotechnology present nomenclature may seem like an arcane art-form Information (NCBI) RefSeqGene project and provides jealously guarded by zealots. -
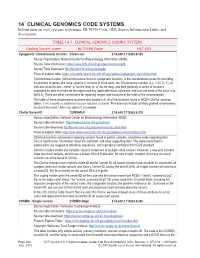
14 CLINICAL GENOMICS CODE SYSTEMS Information on Code Systems with Name, HL70396 Code, OID, Source Information Links, and Description
14 CLINICAL GENOMICS CODE SYSTEMS Information on code systems with name, HL70396 Code, OID, Source Information Links, and description. TABLE 14-1. CLINICAL GENOMICS CODING SYSTEMS Coding System name HL70396 Code HL7 OID Cytogenetic (chromosome) location Chrom-Loc 2.16.840.1.113883.6.335 Source Organization: National Center for Biotechnology Information (NCBI) Source Table Information: https://www.ncbi.nlm.nih.gov/genome/tools/gdp Source Table Download: ftp://ftp.ncbi.nlm.nih.gov/pub/gdp Place to explore table: https://clin-table-search.lhc.nlm.nih.gov/apidoc/cytogenetic_locs/v3/doc.html Chromosome location (AKA chromosome locus or cytogenetic location), is the standardized syntax for recording the position of genes and large variants. It consists of three parts: the Chromosome number (e.g. 1-22, X, Y), an indicator of which arm – either “p” for the short or “q” for the long, and then generally a series of numbers separated by dots that indicate the region and any applicable band, sub-band, and sub-sub-band of the locus (e.g. 2p16.3). There are other conventions for reporting ranges and locations at the ends of the chromosomes. The table of these chromosome locations was loaded with all of the locations found in NCBI’s ClinVar variation tables. It will expand as additional sources become available. This does not include all finely grained chromosome locations that exist. Users can add to it as needed. ClinVar Variant ID CLINVAR-V 2.16.840.1.113883.6.319 Source organization: National Center for Biotechnology Information (NCBI) Source table information: http://www.ncbi.nlm.nih.gov/clinvar/ Source table download: ftp://ftp.ncbi.nlm.nih.gov/pub/clinvar/tab_delimited/ Place to explore table: https://clin-table-search.lhc.nlm.nih.gov/apidoc/variants/v3/doc.html ClinVar processes submissions reporting variants found in patient samples, assertions made regarding their clinical significance, information about the submitter, and other supporting data. -

Haplosaurus Computes Protein Haplotypes for Use in Precision Drug Design
ARTICLE DOI: 10.1038/s41467-018-06542-1 OPEN Haplosaurus computes protein haplotypes for use in precision drug design William Spooner1,2, William McLaren3, Timothy Slidel4, Donna K. Finch4, Robin Butler4, Jamie Campbell4, Laura Eghobamien4, David Rider4, Christine Mione Kiefer 5, Matthew J. Robinson4, Colin Hardman4, Fiona Cunningham 3, Tristan Vaughan4, Paul Flicek 3 & Catherine Chaillan Huntington 4 Selecting the most appropriate protein sequences is critical for precision drug design. Here 1234567890():,; we describe Haplosaurus, a bioinformatic tool for computation of protein haplotypes. Hap- losaurus computes protein haplotypes from pre-existing chromosomally-phased genomic variation data. Integration into the Ensembl resource provides rapid and detailed protein haplotypes retrieval. Using Haplosaurus, we build a database of unique protein haplotypes from the 1000 Genomes dataset reflecting real-world protein sequence variability and their prevalence. For one in seven genes, their most common protein haplotype differs from the reference sequence and a similar number differs on their most common haplotype between human populations. Three case studies show how knowledge of the range of commonly encountered protein forms predicted in populations leads to insights into therapeutic efficacy. Haplosaurus and its associated database is expected to find broad applications in many disciplines using protein sequences and particularly impactful for therapeutics design. 1 Eagle Genomics Ltd., Biodata Innovation Centre, Wellcome Genome Campus, Hinxton, Cambridge CB10 3DR, UK. 2 Genomics England, QMUL Dawson Hall, London EC1M 6BQ, UK. 3 European Molecular Biology Laboratory, European Bioinformatics Institute, Wellcome Genome Campus, Hinxton, Cambridge CB10 1SD, UK. 4 MedImmune Ltd., Granta Park, Cambridge CB21 4QR, UK. 5 MedImmune, Gaithersburg, MD 20878, USA. -

Human Genotype–Phenotype Databases: Aims, Challenges and Opportunities
REVIEWS COMPUTATIONAL TOOLS Human genotype–phenotype databases: aims, challenges and opportunities Anthony J. Brookes1,2 and Peter N. Robinson3–5 Abstract | Genotype–phenotype databases provide information about genetic variation, its consequences and its mechanisms of action for research and health care purposes. Existing databases vary greatly in type, areas of focus and modes of operation. Despite ever larger and more intricate datasets — made possible by advances in DNA sequencing, omics methods and phenotyping technologies — steady progress is being made towards integrating these databases rather than using them as separate entities. The consequential shift in focus from single-gene variants towards large gene panels, exomes, whole genomes and myriad observable characteristics creates new challenges and opportunities in database design, interpretation of variant pathogenicity and modes of data representation and use. The term ‘genotype–phenotype database’ covers a wide gene panels2, whole-exome sequencing (WES)3, clinically range of online and institutional implementations focused WES4 and even whole-genome sequencing5. of systems used for recording and making available However, the power of genomics technologies can be a datasets that include genetic data (for example, DNA double-edged sword: although high-throughput meth- sequences, variants and genotypes), phenotype data ods help find novel disease genes and pathogenic vari- 1Department of Genetics, (observable characteristics of an individual) and corre- ants, they also generate many misleading pathogenicity University of Leicester, lations between the two. In biomedicine, such databases assignments. For instance, a typical WES in a rare dis- Leicester LE1 7RH, UK. 2 are principally focused on human genetic data and the ease medical context will uncover 30,000–100,000 vari- Data to Knowledge for 6 Practice Facility, resulting normal or disease phenotypes. -

Diagnosis of Neurogenetic Disorders Contribution of Next Generation Sequencing and Deep Phenotyping
brain sciences Diagnosis of Neurogenetic Disorders Contribution of Next Generation Sequencing and Deep Phenotyping Edited by Alisdair McNeill Printed Edition of the Special Issue Published in Brain Sciences www.mdpi.com/journal/brainsci Diagnosis of Neurogenetic Disorders Diagnosis of Neurogenetic Disorders: Contribution of Next Generation Sequencing and Deep Phenotyping Special Issue Editor Alisdair McNeill MDPI • Basel • Beijing • Wuhan • Barcelona • Belgrade Special Issue Editor Alisdair McNeill University of Sheffield UK Editorial Office MDPI St. Alban-Anlage 66 4052 Basel, Switzerland This is a reprint of articles from the Special Issue published online in the open access journal Brain Sciences (ISSN 2076-3425) from 2018 to 2019 (available at: https://www.mdpi.com/journal/ brainsci/special issues/diagnosis neurogenetic disorders) For citation purposes, cite each article independently as indicated on the article page online and as indicated below: LastName, A.A.; LastName, B.B.; LastName, C.C. Article Title. Journal Name Year, Article Number, Page Range. ISBN 978-3-03921-610-9 (Pbk) ISBN 978-3-03921-611-6 (PDF) c 2019 by the authors. Articles in this book are Open Access and distributed under the Creative Commons Attribution (CC BY) license, which allows users to download, copy and build upon published articles, as long as the author and publisher are properly credited, which ensures maximum dissemination and a wider impact of our publications. The book as a whole is distributed by MDPI under the terms and conditions of the Creative Commons license CC BY-NC-ND. Contents About the Special Issue Editor ...................................... vii Alisdair McNeill Editorial for Brain Sciences Special Issue: “Diagnosis of Neurogenetic Disorders: Contribution of Next-Generation Sequencing and Deep Phenotyping” Reprinted from: Brain Sci. -

Genes and Variants Underlying Human Congenital Lactic Acidosis—From Genetics to Personalized Treatment
Journal of Clinical Medicine Article Genes and Variants Underlying Human Congenital Lactic Acidosis—From Genetics to Personalized Treatment Irene Bravo-Alonso 1, Rosa Navarrete 1, Ana Isabel Vega 1, Pedro Ruíz-Sala 1, María Teresa García Silva 2, Elena Martín-Hernández 2, Pilar Quijada-Fraile 2, Amaya Belanger-Quintana 3, Sinziana Stanescu 3, María Bueno 4, Isidro Vitoria 5 , Laura Toledo 6, María Luz Couce 7 , Inmaculada García-Jiménez 8, Ricardo Ramos-Ruiz 9 , Miguel Ángel Martín 10 , Lourdes R. Desviat 1, Magdalena Ugarte 1, Celia Pérez-Cerdá 1, Begoña Merinero 1, Belén Pérez 1,* and Pilar Rodríguez-Pombo 1,* 1 Centro de Diagnóstico de Enfermedades Moleculares, Centro de Biología Molecular Severo Ochoa, UAM-CSIC, CIBERER, IDIPAZ, 28049 Madrid, Spain; [email protected] (I.B.-A.); [email protected] (R.N.); [email protected] (A.I.V.); [email protected] (P.R.-S.); [email protected] (L.R.D.); [email protected] (M.U.); [email protected] (C.P.-C.); [email protected] (B.M.) 2 Unidad de Enfermedades Mitocondriales y Enfermedades Metabólicas Hereditarias, Hospital Universitario 12 de Octubre, CIBERER, 28041 Madrid, Spain; [email protected] (M.T.G.S.); [email protected] (E.M.-H.); [email protected] (P.Q.-F.) 3 Unidad de Enfermedades Metabólicas Congénitas, Hospital Universitario Ramón y Cajal, 28034 Madrid, Spain; [email protected] (A.B.-Q.); [email protected] (S.S.) 4 Dpto. de Pediatría, Hospital Universitario Virgen del Rocío, 28034 Sevilla, Spain; -

Algorithms for the Description of Molecular Sequences Issue Date: 2016-12-21
Cover Page The handle http://hdl.handle.net/1887/45045 holds various files of this Leiden University dissertation. Author: Vis, J.K. Title: Algorithms for the description of molecular sequences Issue Date: 2016-12-21 Algorithms for the Description of Molecular Sequences Jonathan K. Vis This publication was supported by the Dutch national program COMMIT. Copyright 2016 by Jonathan K. Vis Copyright Chapter 3: Oxford University Press Copyright Chapter 6: Springer Berlin Heidelberg Open-access: https://openaccess.leidenuniv.nl Typeset using LATEX, figures generated using TIKZ Printed by Ridderprint B.V. ISBN 978-90-9030094-8 Algorithms for the Description of Molecular Sequences Proefschrift ter verkrijging van de graad van Doctor aan de Universiteit Leiden, op gezag van Rector Magnificus prof. mr. C.J.J.M. Stolker, volgens besluit van het College voor Promoties te verdedigen op woensdag 21 december 2016 klokke 11:15 uur door Jonathan Klaas Vis geboren te Leuven, België in 1983 Promotiecommissie Promotores prof.dr. J.N. Kok prof.dr. P.E. Slagboom Copromotor dr. J.F.J. Laros Commissieleden prof.dr.ir. F. Arbab prof.dr. H.J. van den Herik dr. S.D. Olabarriaga Universiteit van Amsterdam prof.dr. A. Plaat dr. P.E.M. Taschner Contents 1 Introduction 11 1.1 Outline . 12 2 Preliminaries 15 2.1 DNA . 16 2.2 RNA . 17 2.3 Proteins . 18 2.4 Transcription . 19 2.5 The genetic code . 20 2.6 Human Genome Variation Society Nomenclature . 22 3 HGVS Description Extraction 25 3.1 Introduction . 26 3.1.1 Transpositions . 27 3.2 Methods . -

The Online Abstract Book
Index Application showcase abstracts 3 Poster abstracts 12 Poster list 94 2 Application showcase abstracts List of participants: 1. Rick de Reuver, Radboud University Nijmegen Medical Centre (UMC St Radboud) VIPtool : a tool for prioritizing pathogenic variants in large annotated sets 2. Egon Willighagen, Maastricht University Bioclipse-OpenTox: interactive predictive toxicology 3. Juha Karjalainen, Department of Genetics, University Medical Center Groningen A gene co-regulation network based on 80,000 samples allows for accurate prediction of gene function 4. Kees Burger/Christine Chichester, Netherlands Bioinformatics Centre A new look for the ConceptWiki 5. Niek Bosch, e-Biogrid/SARA Life Science Grid Portal 6. Kostas Karasavvas, Netherlands Bioinformatics Centre Running Taverna workflows from the web 7. George Beyelas, UMC Groningen Just-enough workflow management system to run bioinformatics analyses on different computational back-ends, such as clusters and grids. 8. Mattias de Hollander, Netherlands Institute of Ecology (NIOO-KNAW) Ongoing efforts of a Galaxy solution for the BiGGrid/SARA HPC Cloud 9. Martijn Vermaat, Leiden University Medical Center Generating and checking complex variant descriptions with Mutalyzer 2 10. Marnix Medema, University of Groningen antiSMASH: genomic analysis of secondary metabolism 11. Pierre-Yves Chibon, Wageningen UR Marker2sequence: From QTLs to potential candidate genes. 12. Jos Lunenberg, Genalice B.V. Genalice: A new groundbreaking Systems Biology Data Processing and Correlation Analysis Platform 13. Farzad Fereidouni, Utrecht University Spectral phasor ImageJ Plugin 3 1. VIPtool : a tool for prioritizing pathogenic variants in large annotated sets Rick de Reuver, Yannick Smits, Nienke Wieskamp, Marcel Nelen, Joris Veltman, Christian Gilissen Department of Human Genetics, Nijmegen Centre for Molecular Life Sciences and Institute for Genetic and Metabolic Disorders, Radboud University Nijmegen Medical Centre, PO Box 9101, 6500 HB Nijmegen, The Netherlands. -

Accurate Mutation Annotation and Functional Prediction Enhance the Applicability of -Omics Data in Precision Medicine Tenghui Chen
Texas Medical Center Library DigitalCommons@The Texas Medical Center UT GSBS Dissertations and Theses (Open Access) Graduate School of Biomedical Sciences 5-2016 Accurate mutation annotation and functional prediction enhance the applicability of -omics data in precision medicine Tenghui Chen Follow this and additional works at: http://digitalcommons.library.tmc.edu/utgsbs_dissertations Part of the Bioinformatics Commons, Computational Biology Commons, Genomics Commons, and the Medicine and Health Sciences Commons Recommended Citation Chen, Tenghui, "Accurate mutation annotation and functional prediction enhance the applicability of -omics data in precision medicine" (2016). UT GSBS Dissertations and Theses (Open Access). Paper 666. This Dissertation (PhD) is brought to you for free and open access by the Graduate School of Biomedical Sciences at DigitalCommons@The exT as Medical Center. It has been accepted for inclusion in UT GSBS Dissertations and Theses (Open Access) by an authorized administrator of DigitalCommons@The exT as Medical Center. For more information, please contact [email protected]. ACCURATE MUTATION ANNOTATION AND FUNCTIONAL PREDICTION ENHANCE THE APPLICABILITY OF -OMICS DATA IN PRECISION MEDICINE by Tenghui Chen, M.S. APPROVED: ___________________________________________ Supervisory Professor: Ken Chen, Ph.D. ______________________________ Keith Baggerly, Ph.D. ______________________________ Roel Verhaak, Ph.D. ______________________________ Han Liang, Ph.D. ______________________________ Marcos Estecio, -

Pathos: a Decision Support System for Reporting High Throughput Sequencing of Cancers in Clinical Diagnostic Laboratories Kenneth D
Doig et al. Genome Medicine (2017) 9:38 DOI 10.1186/s13073-017-0427-z SOFTWARE Open Access PathOS: a decision support system for reporting high throughput sequencing of cancers in clinical diagnostic laboratories Kenneth D. Doig1,2,3,8*, Andrew Fellowes2, Anthony H. Bell2, Andrei Seleznev2, David Ma2, Jason Ellul1, Jason Li1, Maria A. Doyle1, Ella R. Thompson2,3, Amit Kumar1,6,7, Luis Lara1,3, Ravikiran Vedururu2, Gareth Reid2, Thomas Conway1, Anthony T. Papenfuss1,3,5,6 and Stephen B. Fox2,3,4 Abstract Background: The increasing affordability of DNA sequencing has allowed it to be widely deployed in pathology laboratories. However, this has exposed many issues with the analysis and reporting of variants for clinical diagnostic use. Implementing a high-throughput sequencing (NGS) clinical reporting system requires a diverse combination of capabilities, statistical methods to identify variants, global variant databases, a validated bioinformatics pipeline, an auditable laboratory workflow, reproducible clinical assays and quality control monitoring throughout. These capabilities must be packaged in software that integrates the disparate components into a useable system. Results: To meet these needs, we developed a web-based application, PathOS, which takes variant data from a patient sample through to a clinical report. PathOS has been used operationally in the Peter MacCallum Cancer Centre for two years for the analysis, curation and reporting of genetic tests for cancer patients, as well as the curation of large-scale research studies. PathOS has also been deployed in cloud environments allowing multiple institutions to use separate, secure and customisable instances of the system. Increasingly, the bottleneck of variant curation is limiting the adoption of clinical sequencing for molecular diagnostics. -

Distribute! Or! Use! Other! Than! For! Personal! Purposes! Without! The! Permission! Of! Suzanne!Leal!Or!Michael!Nothnagel.!
! ! ! ! ! ! ! ! This! is! copyrighted! material.! Please! do! not! distribute! or! use! other! than! for! personal! purposes! without! the! permission! of! Suzanne!Leal!or!Michael!Nothnagel.! i ii Table&of&Contents& Monday' '................................................................................................................................'1' Introduction*to*finding*pathogenic*variants*using*NGS*data* *..............................................*1* Introduction*to*the*basics* *..................................................................................................................*3* Calculation*of*LOD*scores* *................................................................................................................*13* Cloud*computing* *.................................................................................................................................*20* Tuesday' '.............................................................................................................................'25' Penetrance* *.............................................................................................................................................*25* Software*to*perform*linkage*analysis* *........................................................................................*28* Genotyping*error*detection*using*Merlin* *.................................................................................*33* Homozygosity*mapping* *...................................................................................................................*35*