Pathos: a Decision Support System for Reporting High Throughput Sequencing of Cancers in Clinical Diagnostic Laboratories Kenneth D
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

VISPA: a Computational Pipeline for the Identification and Analysis Of
Calabria et al. Genome Medicine 2014, 6:67 http://genomemedicine.com/content/6/9/67 SOFTWARE Open Access VISPA: a computational pipeline for the identification and analysis of genomic vector integration sites Andrea Calabria1†, Simone Leo2,3†, Fabrizio Benedicenti1, Daniela Cesana1, Giulio Spinozzi1,4, Massimilano Orsini2, Stefania Merella5, Elia Stupka5, Gianluigi Zanetti2 and Eugenio Montini1* Abstract The analysis of the genomic distribution of viral vector genomic integration sites is a key step in hematopoietic stem cell-based gene therapy applications, allowing to assess both the safety and the efficacy of the treatment and to study the basic aspects of hematopoiesis and stem cell biology. Identifying vector integration sites requires ad-hoc bioinformatics tools with stringent requirements in terms of computational efficiency, flexibility, and usability. We developed VISPA (Vector Integration Site Parallel Analysis), a pipeline for automated integration site identification and annotation based on a distributed environment with a simple Galaxy web interface. VISPA was successfully used for the bioinformatics analysis of the follow-up of two lentiviral vector-based hematopoietic stem-cell gene therapy clinical trials. Our pipeline provides a reliable and efficient tool to assess the safety and efficacy of integrating vectors in clinical settings. Background the proximity of the vector’sintegrationsite(IS),a Viral vectors, due to their ability to permanently integrate phenomenon known as insertional mutagenesis (IM) in a target genome, are used to achieve the stable genetic [1,9-12]. The identification of ISs on leukemic cells from modification of therapeutically relevant cells and their GT patients and preclinical models allowed identifying progeny. In particular, γ-retroviral (γ-RVs) and lentiviral the causes of IM and tracking the evolution of the (LVs) vectors are the preferred choice for hematopoietic malignant clone over time [11-16]. -

A Decision Support System for Reporting High Throughput Sequencing of Cancers in Clinical Diagnostic Laboratories Kenneth D
Doig et al. Genome Medicine (2017) 9:38 DOI 10.1186/s13073-017-0427-z SOFTWARE Open Access PathOS: a decision support system for reporting high throughput sequencing of cancers in clinical diagnostic laboratories Kenneth D. Doig1,2,3,8*, Andrew Fellowes2, Anthony H. Bell2, Andrei Seleznev2, David Ma2, Jason Ellul1, Jason Li1, Maria A. Doyle1, Ella R. Thompson2,3, Amit Kumar1,6,7, Luis Lara1,3, Ravikiran Vedururu2, Gareth Reid2, Thomas Conway1, Anthony T. Papenfuss1,3,5,6 and Stephen B. Fox2,3,4 Abstract Background: The increasing affordability of DNA sequencing has allowed it to be widely deployed in pathology laboratories. However, this has exposed many issues with the analysis and reporting of variants for clinical diagnostic use. Implementing a high-throughput sequencing (NGS) clinical reporting system requires a diverse combination of capabilities, statistical methods to identify variants, global variant databases, a validated bioinformatics pipeline, an auditable laboratory workflow, reproducible clinical assays and quality control monitoring throughout. These capabilities must be packaged in software that integrates the disparate components into a useable system. Results: To meet these needs, we developed a web-based application, PathOS, which takes variant data from a patient sample through to a clinical report. PathOS has been used operationally in the Peter MacCallum Cancer Centre for two years for the analysis, curation and reporting of genetic tests for cancer patients, as well as the curation of large-scale research studies. PathOS has also been deployed in cloud environments allowing multiple institutions to use separate, secure and customisable instances of the system. Increasingly, the bottleneck of variant curation is limiting the adoption of clinical sequencing for molecular diagnostics. -

Locus Reference Genomic Sequences: an Improved Basis for Describing Human DNA Variants
Dalgleish et al. Genome Medicine 2010, 2:24 http://genomemedicine.com/content/2/4/24 CORRESPONDENCE Open Access Locus Reference Genomic sequences: an improved basis for describing human DNA variants Raymond Dalgleish1*, Paul Flicek 2, Fiona Cunningham2, Alex Astashyn3, Raymond E Tully3, Glenn Proctor2, Yuan Chen2, William M McLaren2, Pontus Larsson2, Brendan W Vaughan2, Christophe Béroud4, Glen Dobson5, Heikki Lehväslaiho6, Peter EM Taschner7, Johan T den Dunnen7, Andrew Devereau5, Ewan Birney2, Anthony J Brookes1 and Donna R Maglott3 Introduction Abstract In 1993 Ernest Beutler wrote an eloquent letter to the As our knowledge of the complexity of gene editor of the American Journal of Human Genetics high- architecture grows, and we increase our understanding lighting the defi ciencies of the systems then used to of the subtleties of gene expression, the process of describe DNA variants [1]. Th at same year, the editor of accurately describing disease-causing gene variants Human Mutation invited Arthur Beaudet and Lap-Chee has become increasingly problematic. In part, this is Tsui to produce a nomenclature for variants in genes and due to current reference DNA sequence formats that proteins [2]. From these simple beginnings, the last 17 do not fully meet present needs. Here we present the years have borne witness to the steady development of Locus Reference Genomic (LRG) sequence format, the nomenclature used to describe sequence variation which has been designed for the specifi c purpose that is now maintained under the auspices of the Human of gene variant reporting. The format builds on Genome Variation Society (HGVS) [3,4]. To some, the the successful National Center for Biotechnology present nomenclature may seem like an arcane art-form Information (NCBI) RefSeqGene project and provides jealously guarded by zealots. -

INTELLIGENT MEDICINE the Wings of Global Health
INTELLIGENT MEDICINE The Wings of Global Health AUTHOR INFORMATION PACK TABLE OF CONTENTS XXX . • Description p.1 • Editorial Board p.2 • Guide for Authors p.6 ISSN: 2667-1026 DESCRIPTION . Intelligent Medicine is an open access, peer-reviewed journal sponsored and owned by the Chinese Medical Association and designated to publish high-quality research and application in the field of medical-industrial crossover concerning the internet technology, artificial intelligence (AI), data science, medical information, and intelligent devices in the clinical medicine, biomedicine, and public health. Intelligent Medicine appreciates the innovation, pioneering, science, and application, encourages the unique perspectives and suggestions. The topics focus on the computer and data science enabled intelligent medicine, including while not limited to the clinical decision making, computer-assisted surgery, telemedicine, drug development, image analysis and computation, and health management. The journal sets academic columns according to the different disciplines and hotspots. Article types include Research Article: These articles are expected to be original, innovative, and significant, including medical and algorithmic research. The real-world medical research rules and clinical assessment of the usefulness and reliability are recommended for those medical research. The full text is about 6,000 words, with structured abstract of 300 words including Background, Methods, Results, and Conclusion. Editorial: Written by the Editor-in-Chief, Associate Editors, editorial board members, or prestigious invited scientists and policy makers on a broad range of topics from science to policy. Review: Extensive reviews of the recent progress in specific areas of science, involving historical reviews, recent advances made by scientists internationally, and perspective for future development; the full text is about 5,000~6,000 words. -

Developing and Implementing an Institute-Wide Data Sharing Policy Stephanie OM Dyke and Tim JP Hubbard*
Dyke and Hubbard Genome Medicine 2011, 3:60 http://genomemedicine.com/content/3/9/60 CORRESPONDENCE Developing and implementing an institute-wide data sharing policy Stephanie OM Dyke and Tim JP Hubbard* Abstract HapMap Project [7], also decided to follow HGP prac- tices and to share data publicly as a resource for the The Wellcome Trust Sanger Institute has a strong research community before academic publications des- reputation for prepublication data sharing as a result crib ing analyses of the data sets had been prepared of its policy of rapid release of genome sequence (referred to as prepublication data sharing). data and particularly through its contribution to the Following the success of the first phase of the HGP [8] Human Genome Project. The practicalities of broad and of these other projects, the principles of rapid data data sharing remain largely uncharted, especially to release were reaffirmed and endorsed more widely at a cover the wide range of data types currently produced meeting of genomics funders, scientists, public archives by genomic studies and to adequately address and publishers in Fort Lauderdale in 2003 [9]. Meanwhile, ethical issues. This paper describes the processes the Organisation for Economic Co-operation and and challenges involved in implementing a data Develop ment (OECD) Committee on Scientific and sharing policy on an institute-wide scale. This includes Tech nology Policy had established a working group on questions of governance, practical aspects of applying issues of access to research information [10,11], which principles to diverse experimental contexts, building led to a Declaration on access to research data from enabling systems and infrastructure, incentives and public funding [12], and later to a set of OECD guidelines collaborative issues. -

Cancer Genome Interpreter Annotates the Biological and Clinical Relevance of Tumor Alterations David Tamborero1,2, Carlota Rubio-Perez1, Jordi Deu-Pons1,2, Michael P
Tamborero et al. Genome Medicine (2018) 10:25 https://doi.org/10.1186/s13073-018-0531-8 DATABASE Open Access Cancer Genome Interpreter annotates the biological and clinical relevance of tumor alterations David Tamborero1,2, Carlota Rubio-Perez1, Jordi Deu-Pons1,2, Michael P. Schroeder1,3, Ana Vivancos4, Ana Rovira5,6, Ignasi Tusquets5,6,7, Joan Albanell5,6,8, Jordi Rodon4, Josep Tabernero4, Carmen de Torres9, Rodrigo Dienstmann4, Abel Gonzalez-Perez1,2 and Nuria Lopez-Bigas1,2,10* Abstract While tumor genome sequencing has become widely available in clinical and research settings, the interpretation of tumor somatic variants remains an important bottleneck. Here we present the Cancer Genome Interpreter, a versatile platform that automates the interpretation of newly sequenced cancer genomes, annotating the potential of alterations detected in tumors to act as drivers and their possible effect on treatment response. The results are organized in different levels of evidence according to current knowledge, which we envision can support a broad range of oncology use cases. The resource is publicly available at http://www.cancergenomeinterpreter.org. Background to systematically identify genes involved in tumorigen- The accumulation of so-called “driver” genomic alter- esis through the detection of signals of positive selection ations confers on cells tumorigenic capabilities [1]. in their alteration patterns across tumors of some two Thousands of tumor genomes are sequenced around the dozen malignancies [3–6]. However, many of the som- world every year for both research and clinical purposes. atic variants detected in tumors, even those in cancer In some cases the whole genome is sequenced while in genes, still have uncertain significance and thus it is not others the focus is on the exome or a panel of selected clear whether or not they are relevant for tumorigenesis. -

ENVE: a Novel Computational Framework
Varadan et al. Genome Medicine (2015) 7:69 DOI 10.1186/s13073-015-0192-9 METHOD Open Access ENVE: a novel computational framework characterizes copy-number mutational landscapes in colorectal cancers from African American patients Vinay Varadan1,2,7*†, Salendra Singh2, Arman Nosrati3, Lakshmeswari Ravi3, James Lutterbaugh3, Jill S. Barnholtz-Sloan1,2, Sanford D. Markowitz2,3,4,5, Joseph E. Willis2,4,5,6 and Kishore Guda1,2,4,7*† Abstract Reliable detection of somatic copy-number alterations (sCNAs) in tumors using whole-exome sequencing (WES) remains challenging owing to technical (inherent noise) and sample-associated variability in WES data. We present a novel computational framework, ENVE, which models inherent noise in any WES dataset, enabling robust detection of sCNAs across WES platforms. ENVE achieved high concordance with orthogonal sCNA assessments across two colorectal cancer (CRC) WES datasets, and consistently outperformed a best-in-class algorithm, Control-FREEC. We subsequently used ENVE to characterize global sCNA landscapes in African American CRCs, identifying genomic aberrations potentially associated with CRC pathogenesis in this population. ENVE is downloadable at https://github.com/ENVE-Tools/ENVE. Background comprehensively characterize genome-scale DNA alter- Human cancer is caused in part by structural changes ations in tumor tissues. In particular, whole-exome resulting in DNA copy-number alterations at distinct sequencing (WES) offers a cost-effective way of interro- locations in the tumor genome. Identification of such gating mutation and copy-number profiles within somatic copy-number alterations (sCNA) in tumor tis- protein-coding regions in the tumor genome. This has sues has contributed significantly to our understanding resulted in the increasing use of WES in both the re- of the pathogenesis and to the expansion of therapeutic search [8, 9] and clinical settings [10, 11]. -
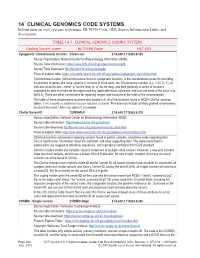
14 CLINICAL GENOMICS CODE SYSTEMS Information on Code Systems with Name, HL70396 Code, OID, Source Information Links, and Description
14 CLINICAL GENOMICS CODE SYSTEMS Information on code systems with name, HL70396 Code, OID, Source Information Links, and description. TABLE 14-1. CLINICAL GENOMICS CODING SYSTEMS Coding System name HL70396 Code HL7 OID Cytogenetic (chromosome) location Chrom-Loc 2.16.840.1.113883.6.335 Source Organization: National Center for Biotechnology Information (NCBI) Source Table Information: https://www.ncbi.nlm.nih.gov/genome/tools/gdp Source Table Download: ftp://ftp.ncbi.nlm.nih.gov/pub/gdp Place to explore table: https://clin-table-search.lhc.nlm.nih.gov/apidoc/cytogenetic_locs/v3/doc.html Chromosome location (AKA chromosome locus or cytogenetic location), is the standardized syntax for recording the position of genes and large variants. It consists of three parts: the Chromosome number (e.g. 1-22, X, Y), an indicator of which arm – either “p” for the short or “q” for the long, and then generally a series of numbers separated by dots that indicate the region and any applicable band, sub-band, and sub-sub-band of the locus (e.g. 2p16.3). There are other conventions for reporting ranges and locations at the ends of the chromosomes. The table of these chromosome locations was loaded with all of the locations found in NCBI’s ClinVar variation tables. It will expand as additional sources become available. This does not include all finely grained chromosome locations that exist. Users can add to it as needed. ClinVar Variant ID CLINVAR-V 2.16.840.1.113883.6.319 Source organization: National Center for Biotechnology Information (NCBI) Source table information: http://www.ncbi.nlm.nih.gov/clinvar/ Source table download: ftp://ftp.ncbi.nlm.nih.gov/pub/clinvar/tab_delimited/ Place to explore table: https://clin-table-search.lhc.nlm.nih.gov/apidoc/variants/v3/doc.html ClinVar processes submissions reporting variants found in patient samples, assertions made regarding their clinical significance, information about the submitter, and other supporting data. -

A 26-Hour System of Highly Sensitive Whole Genome Sequencing for Emergency Management of Genetic Diseases Neil A
Miller et al. Genome Medicine (2015) 7:100 DOI 10.1186/s13073-015-0221-8 METHOD Open Access A 26-hour system of highly sensitive whole genome sequencing for emergency management of genetic diseases Neil A. Miller1†, Emily G. Farrow1,2,3,4†, Margaret Gibson1, Laurel K. Willig1,2,4, Greyson Twist1, Byunggil Yoo1, Tyler Marrs1, Shane Corder1, Lisa Krivohlavek1, Adam Walter1, Josh E. Petrikin1,2,4, Carol J. Saunders1,2,3,4, Isabelle Thiffault1,3, Sarah E. Soden1,2,4, Laurie D. Smith1,2,3,4, Darrell L. Dinwiddie5, Suzanne Herd1, Julie A. Cakici1, Severine Catreux6, Mike Ruehle6 and Stephen F. Kingsmore1,2,3,4,7* Abstract While the cost of whole genome sequencing (WGS) is approaching the realm of routine medical tests, it remains too tardy to help guide the management of many acute medical conditions. Rapid WGS is imperative in light of growing evidence of its utility in acute care, such as in diagnosis of genetic diseases in very ill infants, and genotype-guided choice of chemotherapy at cancer relapse. In such situations, delayed, empiric, or phenotype-based clinical decisions may meet with substantial morbidity or mortality. We previously described a rapid WGS method, STATseq, with a sensitivity of >96 % for nucleotide variants that allowed a provisional diagnosis of a genetic disease in 50 h. Here improvements in sequencing run time, read alignment, and variant calling are described that enable 26-h time to provisional molecular diagnosis with >99.5 % sensitivity and specificity of genotypes. STATseq appears to be an appropriate strategy for acutely ill patients with potentially actionable genetic diseases. -

Exploration of Haplotype Research Consortium Imputation for Genome-Wide Association Studies in 20,032 Generation Scotland Participants Reka Nagy1, Thibaud S
Nagy et al. Genome Medicine (2017) 9:23 DOI 10.1186/s13073-017-0414-4 RESEARCH Open Access Exploration of haplotype research consortium imputation for genome-wide association studies in 20,032 Generation Scotland participants Reka Nagy1, Thibaud S. Boutin1, Jonathan Marten1, Jennifer E. Huffman1, Shona M. Kerr1, Archie Campbell2, Louise Evenden3, Jude Gibson3, Carmen Amador1, David M. Howard4, Pau Navarro1, Andrew Morris5, Ian J. Deary6, Lynne J. Hocking7, Sandosh Padmanabhan8, Blair H. Smith9, Peter Joshi10, James F. Wilson10, Nicholas D. Hastie1, Alan F. Wright1, Andrew M. McIntosh4,6, David J. Porteous2,6, Chris S. Haley1, Veronique Vitart1 and Caroline Hayward1* Abstract Background: The Generation Scotland: Scottish Family Health Study (GS:SFHS) is a family-based population cohort with DNA, biological samples, socio-demographic, psychological and clinical data from approximately 24,000 adult volunteers across Scotland. Although data collection was cross-sectional, GS:SFHS became a prospective cohort due to of the ability to link to routine Electronic Health Record (EHR) data. Over 20,000 participants were selected for genotyping using a large genome-wide array. Methods: GS:SFHS was analysed using genome-wide association studies (GWAS) to test the effects of a large spectrum of variants, imputed using the Haplotype Research Consortium (HRC) dataset, on medically relevant traits measured directly or obtained from EHRs. The HRC dataset is the largest available haplotype reference panel for imputation of variants in populations of European ancestry and allows investigation of variants with low minor allele frequencies within the entire GS:SFHS genotyped cohort. Results: Genome-wide associations were run on 20,032 individuals using both genotyped and HRC imputed data. -

Omic Personality: Implications of Stable
Tabassum et al. Genome Medicine (2015) 7:88 DOI 10.1186/s13073-015-0209-4 RESEARCH Open Access Omic personality: implications of stable transcript and methylation profiles for personalized medicine Rubina Tabassum1,2, Ambily Sivadas1,3, Vartika Agrawal1,4, Haozheng Tian1, Dalia Arafat1 and Greg Gibson1* Abstract Background: Personalized medicine is predicated on the notion that individual biochemical and genomic profiles are relatively constant in times of good health and to some extent predictive of disease or therapeutic response. We report a pilot study quantifying gene expression and methylation profile consistency over time, addressing the reasons for individual uniqueness, and its relation to N = 1 phenotypes. Methods: Whole blood samples from four African American women, four Caucasian women, and four Caucasian men drawn from the Atlanta Center for Health Discovery and Well Being study at three successive 6-month intervals were profiled by RNA-Seq, miRNA-Seq, and Illumina Methylation 450 K arrays. Standard regression approaches were used to evaluate the proportion of variance for each type of omic measure among individuals, and to quantify correlations among measures and with clinical attributes related to wellness. Results: Longitudinal omic profiles were in general highly consistent over time, with an average of 67 % variance in transcript abundance, 42 % in CpG methylation level (but 88 % for the most differentiated CpG per gene), and 50 % in miRNA abundance among individuals, which are all comparable to 74 % variance among individuals for 74 clinical traits. One third of the variance could be attributed to differential blood cell type abundance, which was also fairly stable over time, and a lesser amount to expression quantitative trait loci (eQTL) effects. -
![Downloaded from the CAVA Integration with Data Generated by Pre-NGS Methods Webpage [19]](https://docslib.b-cdn.net/cover/5130/downloaded-from-the-cava-integration-with-data-generated-by-pre-ngs-methods-webpage-19-2125130.webp)
Downloaded from the CAVA Integration with Data Generated by Pre-NGS Methods Webpage [19]
Münz et al. Genome Medicine (2015) 7:76 DOI 10.1186/s13073-015-0195-6 METHOD Open Access CSN and CAVA: variant annotation tools for rapid, robust next-generation sequencing analysis in the clinical setting Márton Münz1†, Elise Ruark2†, Anthony Renwick2, Emma Ramsay2, Matthew Clarke2, Shazia Mahamdallie2,3, Victoria Cloke3, Sheila Seal2,3, Ann Strydom2,3, Gerton Lunter1 and Nazneen Rahman2,3,4* Abstract Background: Next-generation sequencing (NGS) offers unprecedented opportunities to expand clinical genomics. It also presents challenges with respect to integration with data from other sequencing methods and historical data. Provision of consistent, clinically applicable variant annotation of NGS data has proved difficult, particularly of indels, an important variant class in clinical genomics. Annotation in relation to a reference genome sequence, the DNA strand of coding transcripts and potential alternative variant representations has not been well addressed. Here we present tools that address these challenges to provide rapid, standardized, clinically appropriate annotation of NGS data in line with existing clinical standards. Methods: We developed a clinical sequencing nomenclature (CSN), a fixed variant annotation consistent with the principles of the Human Genome Variation Society (HGVS) guidelines, optimized for automated variant annotation of NGS data. To deliver high-throughput CSN annotation we created CAVA (Clinical Annotation of VAriants), a fast, lightweight tool designed for easy incorporation into NGS pipelines. CAVA allows transcript specification, appropriately accommodates the strand of a gene transcript and flags variants with alternative annotations to facilitate clinical interpretation and comparison with other datasets. We evaluated CAVA in exome data and a clinical BRCA1/BRCA2 gene testing pipeline.