Calcium Signaling in Cardiac Myocytes
Total Page:16
File Type:pdf, Size:1020Kb

Load more
Recommended publications
-

Unnatural Verticilide Enantiomer Inhibits Type 2 Ryanodine Receptor-Mediated Calcium Leak and Is Antiarrhythmic
Unnatural verticilide enantiomer inhibits type 2 ryanodine receptor-mediated calcium leak and is antiarrhythmic Suzanne M. Batistea,1, Daniel J. Blackwellb,1, Kyungsoo Kimb,1, Dmytro O. Kryshtalb, Nieves Gomez-Hurtadob, Robyn T. Rebbeckc, Razvan L. Corneac, Jeffrey N. Johnstona,2, and Bjorn C. Knollmannb,2 aDepartment of Chemistry, Vanderbilt University, Nashville, TN 37235; bDepartment of Medicine, Vanderbilt University Medical Center, Nashville, TN 37232; and cDepartment of Biochemistry, Molecular Biology, and Biophysics, University of Minnesota, Minneapolis, MN 55455 Edited by Dale L. Boger, The Scripps Research Institute, La Jolla, CA, and approved January 15, 2019 (received for review September 27, 2018) Ca2+ leak via ryanodine receptor type 2 (RyR2) can cause poten- heart diseases associated with both atrial and ventricular arrhyth- tially fatal arrhythmias in a variety of heart diseases and has also mia (9). Mutations in RyR2 and its binding partners, which increase + been implicated in neurodegenerative and seizure disorders, mak- SR Ca2 leak, cause primary atrial and ventricular arrhythmia ing RyR2 an attractive therapeutic target for drug development. syndromes such as catecholaminergic polymorphic ventricular Here we synthesized and investigated the fungal natural product tachycardia (CPVT), providing strong evidence for the mechanistic and known insect RyR antagonist (−)-verticilide and several conge- contribution of RyR2 to arrhythmia risk in humans (10). Further ners to determine their activity against mammalian RyR2. Although support comes from gene-targeted mouse models of CPVT, where + the cyclooligomeric depsipeptide natural product (−)-verticilide had catecholamine-induced spontaneous Ca2 release from the SR no effect, its nonnatural enantiomer [ent-(+)-verticilide] signifi- via RyR2 generates potentially fatal cardiac arrhythmias (11, 12). -

Calcium-Induced Calcium Release in Smooth Muscle7 Loose Coupling
View metadata, citation and similar papers at core.ac.uk brought to you by CORE provided by PubMed Central Calcium-induced Calcium Release in Smooth Muscle✪ Loose Coupling between the Action Potential and Calcium Release M.L. Collier, G. Ji, Y.-X. Wang, and M.I. Kotlikoff From the Department of Animal Biology, School of Veterinary Medicine, University of Pennsylvania, Philadelphia, Pennsylvania 19104-6046 abstract Calcium-induced calcium release (CICR) has been observed in cardiac myocytes as elementary cal- cium release events (calcium sparks) associated with the opening of L-type Ca2ϩ channels. In heart cells, a tight coupling between the gating of single L-type Ca2ϩ channels and ryanodine receptors (RYRs) underlies calcium re- lease. Here we demonstrate that L-type Ca2ϩ channels activate RYRs to produce CICR in smooth muscle cells in the form of Ca2ϩ sparks and propagated Ca2ϩ waves. However, unlike CICR in cardiac muscle, RYR channel open- ing is not tightly linked to the gating of L-type Ca2ϩ channels. L-type Ca2ϩ channels can open without triggering Ca2ϩ sparks and triggered Ca2ϩ sparks are often observed after channel closure. CICR is a function of the net flux of Ca2ϩ ions into the cytosol, rather than the single channel amplitude of L-type Ca2ϩ channels. Moreover, unlike CICR in striated muscle, calcium release is completely eliminated by cytosolic calcium buffering. Thus, L-type Ca2ϩ channels are loosely coupled to RYR through an increase in global [Ca2ϩ] due to an increase in the effective distance between L-type Ca2ϩ channels and RYR, resulting in an uncoupling of the obligate relationship that exists in striated muscle between the action potential and calcium release. -

Microarchitecture of the Dyad
Cardiovascular Research (2013) 98, 169–176 SPOTLIGHT REVIEW doi:10.1093/cvr/cvt025 Microarchitecture of the dyad David R.L. Scriven, Parisa Asghari, and Edwin D.W. Moore* Department of Cellular and Physiological Sciences, Life Sciences Institute, University of British Columbia, 2350 Health Sciences Mall, Vancouver, BC, Canada V6T 1Z3 Received 12 December 2012; revised 2 February 2013; accepted 4 February 2013; online publish-ahead-of-print 11 February 2013 Downloaded from https://academic.oup.com/cardiovascres/article/98/2/169/278625 by guest on 23 September 2021 Abstract This review highlights recent and ongoing discoveries that are transforming the previously held view of dyad structure and function. New data show that dyads vary greatly in both structure and in their associated molecules. Dyads can contain varying numbers of type 2 ryanodine receptor (RYR2) clusters that range in size from one to hundreds of tetramers and they can adopt numerous orientations other than the expected checkerboard. The association of Cav1.2 with RYR2, which defines the couplon, is not absolute, leading to a number of scenarios such as dyads without couplons and those in which only a fraction of the clusters are in couplons. Different dyads also vary in the transporters and exchangers with which they are associated producing functional differences that amplify their structural diversity. The essential role of proteins, such as junctophilin-2, calsequestrin, triadin, and junctin that main- tain both the functional and structural integrity of the dyad have recently been elucidated giving a new mechanistic understanding of heart diseases, such as arrhythmias, hypertension, failure, and sudden cardiac death. -

Cardiovascular Physiology
CARDIOVASCULAR PHYSIOLOGY Ida Sletteng Karlsen • Trym Reiberg Second Edition 15 March 2020 Copyright StudyAid 2020 Authors Trym Reiberg Ida Sletteng Karlsen Illustrators Nora Charlotte Sønstebø Ida Marie Lisle Amalie Misund Ida Sletteng Karlsen Trym Reiberg Booklet Disclaimer All rights reserved. No part oF this book may be reproduced in any Form on by an electronic or mechanical means, without permission From StudyAid. Although the authors have made every efFort to ensure the inFormation in the booklet was correct at date of publishing, the authors do not assume and hereby disclaim any liability to any part For any inFormation that is omitted or possible errors. The material is taken From a variety of academic sources as well as physiology lecturers, but are Further incorporated and summarized in an original manner. It is important to note, the material has not been approved by professors of physiology. All illustrations in the booklet are original. This booklet is made especially For students at the Jagiellonian University in Krakow by tutors in the StudyAid group (students at JU). It is available as a PDF and is available for printing. If you have any questions concerning copyrights oF the booklet please contact [email protected]. About StudyAid StudyAid is a student organization at the Jagiellonian University in Krakow. Throughout the academic year we host seminars in the major theoretical subjects: anatomy, physiology, biochemistry, immunology, pathophysiology, supplementing the lectures provided by the university. We are a group of 25 tutors, who are students at JU, each with their own Field oF specialty. To make our seminars as useful and relevant as possible, we teach in an interactive manner often using drawings and diagrams to help students remember the concepts. -

Non-Coding Rnas in the Cardiac Action Potential and Their Impact on Arrhythmogenic Cardiac Diseases
Review Non-Coding RNAs in the Cardiac Action Potential and Their Impact on Arrhythmogenic Cardiac Diseases Estefania Lozano-Velasco 1,2 , Amelia Aranega 1,2 and Diego Franco 1,2,* 1 Cardiovascular Development Group, Department of Experimental Biology, University of Jaén, 23071 Jaén, Spain; [email protected] (E.L.-V.); [email protected] (A.A.) 2 Fundación Medina, 18016 Granada, Spain * Correspondence: [email protected] Abstract: Cardiac arrhythmias are prevalent among humans across all age ranges, affecting millions of people worldwide. While cardiac arrhythmias vary widely in their clinical presentation, they possess shared complex electrophysiologic properties at cellular level that have not been fully studied. Over the last decade, our current understanding of the functional roles of non-coding RNAs have progressively increased. microRNAs represent the most studied type of small ncRNAs and it has been demonstrated that miRNAs play essential roles in multiple biological contexts, including normal development and diseases. In this review, we provide a comprehensive analysis of the functional contribution of non-coding RNAs, primarily microRNAs, to the normal configuration of the cardiac action potential, as well as their association to distinct types of arrhythmogenic cardiac diseases. Keywords: cardiac arrhythmia; microRNAs; lncRNAs; cardiac action potential Citation: Lozano-Velasco, E.; Aranega, A.; Franco, D. Non-Coding RNAs in the Cardiac Action Potential 1. The Electrical Components of the Adult Heart and Their Impact on Arrhythmogenic The adult heart is a four-chambered organ that propels oxygenated blood to the entire Cardiac Diseases. Hearts 2021, 2, body. It is composed of atrial and ventricular chambers, each of them with distinct left and 307–330. -

Calcium Signaling at the Endoplasmic Reticulum Fine-Tuning Stress
Cell Calcium 70 (2018) 24–31 Contents lists available at ScienceDirect Cell Calcium journal homepage: www.elsevier.com/locate/ceca Review Calcium signaling at the endoplasmic reticulum: fine-tuning stress responses T ⁎ Amado Carreras-Suredaa,b,c, Philippe Pihána,b,c, Claudio Hetza,b,c,d,e, a Center for Geroscience, Brain Health and Metabolism, Faculty of Medicine, University of Chile, Chile b Biomedical Neuroscience Institute, Faculty of Medicine, University of Chile, Santiago, Chile c Program of Cellular and Molecular Biology, Institute of Biomedical Sciences, University of Chile, Santiago, Chile d Buck Institute for Research on Aging, Novato, CA, 94945, USA e Department of Immunology and Infectious Diseases, Harvard School of Public Health, Boston, MA 02115, USA ARTICLE INFO ABSTRACT Keywords: Endoplasmic reticulum (ER) calcium signaling is implicated in a myriad of coordinated cellular processes. The ER homeostasis ER calcium content is tightly regulated as it allows a favorable environment for protein folding, in addition to ER stress operate as a major reservoir for fast and specific release of calcium. Altered ER homeostasis impacts protein Calcium handling mechanisms folding, activating the unfolded protein response (UPR) as a rescue mechanism to restore proteostasis. ER cal- Calcium homeostasis cium release impacts mitochondrial metabolism and also fine-tunes the threshold to undergo apoptosis under Unfolded protein response chronic stress. The global coordination between UPR signaling and energetic demands takes place at mi- Mitochondrial associated membranes Mitochondria biology tochondrial associated membranes (MAMs), specialized subdomains mediating interorganelle communication. Here we discuss current models explaining the functional relationship between ER homeostasis and various cellular responses to coordinate proteostasis and metabolic maintenance. -

Calcium Signaling in Aging and Neurodegenerative Diseases 2019
International Journal of Molecular Sciences Meeting Report Calcium Signaling in Aging and Neurodegenerative Diseases 2019 Luísa Cortes 1,2 , João Malva 2,3,4, Ana Cristina Rego 1,2,3 and Cláudia F. Pereira 1,2,3,* 1 Center for Neuroscience and Cell Biology (CNC), University of Coimbra, Rua Larga, Faculty of Medicine, Polo I, 1st floor, 3004-504 Coimbra, Portugal; [email protected] (L.C.); [email protected] (A.C.R.) 2 CIBB-Center for Innovative Biomedicine and Biotechnology, University of Coimbra, Rua Larga, Faculty of Medicine, Polo I, 1st floor, 3004-504 Coimbra, Portugal; [email protected] 3 Faculty of Medicine, Azinhaga de Santa Comba, Celas, 3000-548 Coimbra, Portugal 4 iCRB- Coimbra Institute for Clinical and Biomedical Research; Azinhaga de Santa Comba, Celas, 3000-548 Coimbra, Portugal * Correspondence: [email protected] Received: 28 December 2019; Accepted: 4 February 2020; Published: 7 February 2020 Abstract: The European Calcium Society (ECS) workshop, which is held every 2 years, is a dedicated meeting of scientists interested in the elucidation of the action of calcium binding, calcium signaling and the study of proteins and organelles, such as mitochondria and endoplasmic reticulum, thereby involved, either in health and disease conditions. The 8th edition of the ECS workshop was organized by a group of researchers from the University of Coimbra, Portugal, in close collaboration with ECS board members. Thanks to the central role of “Calcium Signaling in Aging and Neurodegenerative Disorders”, the ECS 2019 workshop was attended by 62 experts who presented their results in a plenary lecture and five regular symposia, two oral communication sessions and two poster sessions, followed by a hands-on session on calcium imaging. -

Electrical Activity of the Heart: Action Potential, Automaticity, and Conduction 1 & 2 Clive M
Electrical Activity of the Heart: Action Potential, Automaticity, and Conduction 1 & 2 Clive M. Baumgarten, Ph.D. OBJECTIVES: 1. Describe the basic characteristics of cardiac electrical activity and the spread of the action potential through the heart 2. Compare the characteristics of action potentials in different parts of the heart 3. Describe how serum K modulates resting potential 4. Describe the ionic basis for the cardiac action potential and changes in ion currents during each phase of the action potential 5. Identify differences in electrical activity across the tissues of the heart 6. Describe the basis for normal automaticity 7. Describe the basis for excitability 8. Describe the basis for conduction of the cardiac action potential 9. Describe how the responsiveness relationship and the Na+ channel cycle modulate cardiac electrical activity I. BASIC ELECTROPHYSIOLOGIC CHARACTERISTICS OF CARDIAC MUSCLE A. Electrical activity is myogenic, i.e., it originates in the heart. The heart is an electrical syncitium (i.e., behaves as if one cell). The action potential spreads from cell-to-cell initiating contraction. Cardiac electrical activity is modulated by the autonomic nervous system. B. Cardiac cells are electrically coupled by low resistance conducting pathways gap junctions located at the intercalated disc, at the ends of cells, and at nexus, points of side-to-side contact. The low resistance pathways (wide channels) are formed by connexins. Connexins permit the flow of current and the spread of the action potential from cell-to-cell. C. Action potentials are much longer in duration in cardiac muscle (up to 400 msec) than in nerve or skeletal muscle (~5 msec). -

Calcium Signaling and Cardiac Arrhythmias
Review Calcium Signaling Series Donald M. Bers, Guest Editor Calcium Signaling and Cardiac Arrhythmias Andrew P. Landstrom, Dobromir Dobrev, Xander H.T. Wehrens Abstract: There has been a significant progress in our understanding of the molecular mechanisms by which calcium (Ca2+) ions mediate various types of cardiac arrhythmias. A growing list of inherited gene defects can cause potentially lethal cardiac arrhythmia syndromes, including catecholaminergic polymorphic ventricular Downloaded from tachycardia, congenital long QT syndrome, and hypertrophic cardiomyopathy. In addition, acquired deficits of multiple Ca2+-handling proteins can contribute to the pathogenesis of arrhythmias in patients with various types of heart disease. In this review article, we will first review the key role of Ca2+ in normal cardiac function—in particular, excitation–contraction coupling and normal electric rhythms. The functional involvement of Ca2+ in distinct arrhythmia mechanisms will be discussed, followed by various inherited arrhythmia syndromes caused 2+ http://circres.ahajournals.org/ by mutations in Ca -handling proteins. Finally, we will discuss how changes in the expression of regulation of Ca2+ channels and transporters can cause acquired arrhythmias, and how these mechanisms might be targeted for therapeutic purposes. (Circ Res. 2017;120:1969-1993. DOI: 10.1161/CIRCRESAHA.117.310083.) Key Words: arrhythmias, cardiac ■ atrial fibrillation ■ calcium channels ■ cardiomyopathy ■ ryanodine receptor calcium release channel 2+ by guest on June 11, 2017 he bivalent cation calcium (Ca ) represents one of the Overview of Excitation–Contraction Tmost ubiquitous signal transduction molecules known.1 Coupling in the Heart It mediates a diverse array of biological functions including Regular contraction of the heart requires the conversion of muscle contraction, cellular exocytosis, neuronal activity, and electric activation (excitation) into mechanical force (con- triggering of programmed cell death. -
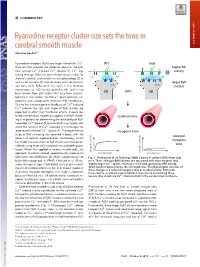
Ryanodine Receptor Cluster Size Sets the Tone in Cerebral Smooth Muscle
COMMENTARY Ryanodine receptor cluster size sets the tone in cerebral smooth muscle COMMENTARY Christian Soellera,1 + Ryanodine receptors (RyRs) are large intracellular Ca2 WT mdx channels that provide the molecular basis of the pro- K+ K+ higher BK + + + cess termed Ca2 -induced Ca2 release (1). Ca2 sig- activity naling through RyRs has been shown to be critical for CaC BK BK skeletal, cardiac, and smooth muscle physiology (2) as well as for neurons (3) and secretory cells like pancre- larger RyR atic beta cells. RyRs were first seen in the electron RyRs RyRs clusters microscope as ∼30-nm-size particles (4), and it had SMCs been known from EM studies that they form clusters, SR SR typically in the narrow “junctional” space between sar- colemma and sarcoplasmic reticulum (SR) membranes. + Due to the inherent positive feedback of Ca2 -induced + Ca2 release, the size and shape of RyR clusters are expected to affect their functional activity. Indeed, de- SMCs SMCs tailed mathematical modeling suggests that RyR cluster- cerebral artery ing is important for determining the excitability of RyR- + mediated Ca2 release (5, 6) and could, in principle, also + affect the amount of Ca2 released in microscopic re- 2+ lease events termed Ca sparks (7). The experimental myogenic tone study of RyR clustering has received a boost with the reduced advent of optical superresolution microscopy, which myogenic has made the assessment of RyR cluster size more acces- sible by using essentially standard immunolabeling pro- tone tocols. When first applied in cardiac muscle cells, this myogenic tone (%) tone myogenic approach revealed a broad, approximately exponential vessel pressure (%) tone myogenic vessel pressure RyR cluster size distribution (8), which had previously not Fig. -

Two Components of the Cardiac Action Potential I
Two Components of the Cardiac Action Potential I. Voltage-time course and the effect of acet.flcholine on atrial and nodal cells of the rabbit heart ANTONIO PAES de CARVALHO, BRIAN FRANCIS HOFFMAN, and MARILENE de PAULA CARVALHO From the Instituto de Biofisica da Universidade Federal do Rio de Janeiro, Rio de Janeiro (GB), Brazil, and the Department of Pharmacology, College of Physicians and Surgeons, Columbia University, New York 10032 ABSTRACT Transmembrane potentials recorded from the rabbit heart in vitro were displayed as voltage against time (V, t display), and dV/dt against voltage (I?, V or phase-plane display). Acetylcholine was applied to the record- ing site by means of a hydraulic system. Results showed that (a) differences in time course of action potential upstroke can be explained in terms of the rela- tive magnitude of fast and slow phases of depolarization; (b) acetylcholine is capable of depressing the slow phase of depolarization as well as the plateau of the action potential; and (¢) action potentials from nodal (SA and AV) cells seem to lack the initial fast phase. These results were construed to support a two-component hypothesis for cardiac electrogenesis. The hypothesis states that cardiac action potentials are composed of two distinct and physiologically separable "components" which result from discrete mechanisms. An initial fast component is a sodium spike similar to that of squid nerve. The slow com- ponent, which accounts for both a slow depolarization during phase 0 and the plateau, probably is dependent on the properties of a slow inward current hav- ing a positive equilibrium potential, coupled to a decrease in the resting potas- sium conductance. -

Are Ryanodine Receptors Important for Diastolic Depolarization in Heart?
ARE RYANODINE RECEPTORS IMPORTANT FOR DIASTOLIC DEPOLARIZATION IN HEART? A Dissertation submitted in partial fulfillment of the requirement for the Degree of Doctor of Medicine in Physiology (Branch – V) Of The Tamilnadu Dr. M.G.R Medical University, Chennai -600 032 Department of Physiology Christian Medical College, Vellore Tamilnadu April 2017 Ref: …………. Date: …………. CERTIFICATE This is to certify that the thesis entitled “Are ryanodine receptors important for diastolic depolarization in heart?” is a bonafide, original work carried out by Dr.Teena Maria Jose , in partial fulfillment of the rules and regulations for the M.D – Branch V Physiology examination of the Tamilnadu Dr. M.G.R. Medical University, Chennai to be held in April- 2017. Dr. Sathya Subramani, Professor and Head Department of Physiology, Christian Medical College, Vellore – 632 002 Ref: …………. Date: …………. CERTIFICATE This is to certify that the thesis entitled “Are ryanodine receptors important for diastolic depolarization in heart?” is a bonafide, original work carried out by Dr.Teena Maria Jose , in partial fulfillment of the rules and regulations for the M.D – Branch V Physiology examination of the Tamilnadu Dr. M.G.R. Medical University, Chennai to be held in April- 2017. Dr. Anna B Pulimood, Principal, Christian Medical College, Vellore – 632 002 DECLARATION I hereby declare that the investigations that form the subject matter for the thesis entitled “Are ryanodine receptors important for diastolic depolarization in heart?” were carried out by me during my term as a post graduate student in the Department of Physiology, Christian Medical College, Vellore. This thesis has not been submitted in part or full to any other university.