Large-Scale Screening of Thalassemia in Ji'an, P.R. China
Total Page:16
File Type:pdf, Size:1020Kb

Load more
Recommended publications
-

Amphibia: Anura: Megophryidae) from Mount Jinggang, China, Based on Molecular and Morphological Data
Zootaxa 3546: 53–67 (2012) ISSN 1175-5326 (print edition) www.mapress.com/zootaxa/ ZOOTAXA Copyright © 2012 · Magnolia Press Article ISSN 1175-5334 (online edition) urn:lsid:zoobank.org:pub:94669404-4465-48A9-AB35-8860F1E46C82 Description of a new species of the genus Xenophrys Günther, 1864 (Amphibia: Anura: Megophryidae) from Mount Jinggang, China, based on molecular and morphological data YING-YONG WANG1,4, TIAN-DU ZHANG1, JIAN ZHAO2, YIK-HEI SUNG3, JIAN-HUAN YANG1, HONG PANG1 & ZHONG ZHANG2 1State Key Laboratory of Biocontrol / The Museum of Biology, School of Life Sciences, Sun Yat-sen University, Guangzhou 510275, P. R . C h in a 2Jinggangshan National Nature Reserve, Ciping, 343600, Jinggangshan City, Jiangxi, P.R. China 3Kadoorie Conservation China, Kadoorie Farm and Botanic Garden, Lam Kam Road, Tai Po, Hong Kong 4Corresponding author. E-mail: [email protected] Abstract A new species, Xenophrys jinggangensis sp. nov., is described based on a series of specimens collected from Mount Jing- gang, Jiangxi Province, Eastern China. The new species can be easily distinguished from other known congeners by mor- phology, morphometrics and molecular data of the mitochondrial 16SrRNA gene. The new species is characterized by its small size with adult females measuring 38.4–41.6 mm in snout-vent length and males measuring 35.1–36.7 mm; head length approximately equal to head width; tympanum large and distinct, about 0.8 times of eye diameter; vomerine teeth on two weak ridges; tongue not notched behind; relative finger length II < I < IV < III; slight lateral fringes present on digits; toes bases with thick, fleshy web; dorsum with tubercles and swollen dorsolateral folds; large pustules scattered on flanks; and unique color patterns. -

RESETTLEMENT PLAN of Shihutang Hydropower Project on Ganjiang River in Jiangxi Province Public Disclosure Authorized Public Disclosure Authorized
RP617 Public Disclosure Authorized RESETTLEMENT PLAN of Shihutang Hydropower Project on Ganjiang River in Jiangxi Province Public Disclosure Authorized Public Disclosure Authorized China Pearl River Water Resources Planning, Design and Survey Co. Ltd. Jiangxi Provincial Water Conservancy Planning and Designing Institute Public Disclosure Authorized Feb 2008 Authofized: LI Xue-ning Checked & Ratified: HUANG You-sheng Examined: LI Chang-sun Verified: MENG Chao-hui HU Jian-jun Editor: WAN Hai-ping TU Lan-tao XUE Bin Attendee: ZHOU Xiao-hua YOU Qin-sheng FENG Chang-jing CHENG Shi-yan WAN Lu-jian ZHANG Zi-lin LIU Qi-jun Contents PURPOSES OF RESETTLEMENT PLAN AND DEFINITION FOR RELOCATION................1 1 REPORT GENERAL.........................................................................................................................4 1.1 Project background........................................................................................................................... 4 1.2 Project general.................................................................................................................................. 5 1.3 Project impact................................................................................................................................... 6 1.4 Policy framework of resettlement relocation ................................................................................... 8 1.5 Implementation planning of resettlement relocation....................................................................... -

45022-002: Jiangxi Ji'an Sustainable Urban Transport Project
Social Monitoring Report Project Number: 45022-002 Semi-Annual Report May 2018 PRC: Jiangxi Ji’an Sustainable Urban Transport Project Prepared by Jiangxi Academy of Social Science for the People’s Republic of China and the Asian Development Bank. This social monitoring report is a document of the borrower. The views expressed herein do not necessarily represent those of ADB's Board of Directors, Management, or staff, and may be preliminary in nature. In preparing any country program or strategy, financing any project, or by making any designation of or reference to a particular territory or geographic area in this document, the Asian Development Bank does not intend to make any judgments as to the legal or other status of any territory or area. Asian Development Bank 3216-PRC ADB Loan Ji’an Urban Sustainable Transport Project External Social and Resettlement Monitoring No.2 Report (October 2017 to March 2018) Monitoring agency: Jiangxi Academy of Social Science May 2018 Executive Abstract According to the ADB’s requirement, the external monitoring of resettlement will be carried out once every six months during the resettlement implementation. The team of EM carried out a monitoring and evaluation on implementation course of LA, HD and resettlement from October 2017 to March 2018. The team adopted document method, sampling survey and depth interview method (including interview with affected households and heads of EA.) The results of E&M show both five roads involving LA and HD. The expropriated land and housing carried out based on state policies, and met with the standards of resettlement plan approved by ADB. -

County, Province 包装厂中文名chinese Name of Packing House
序号 注册登记号 所在地 Location: 包装厂中文名 包装厂英文名 包装厂中文地址 包装厂英文地址 Numbe Registered Location County, Province Chinese Name of Packing house English Name of Packing house Address in Chinese Address in English r Number 1 北京平谷 PINGGU,BEIJING 北京凤凰山投资管理中心 BEIJING FENGHUANGSHAN INVESTMENT MANAGEMENT CENTER 平谷区峪口镇 YUKOU,PINGU DISTRICT,BEIJING 1100GC001 2 北京平谷 PINGGU,BEIJING 北京东四道岭果品产销专业合作社 BEIJING DONGSIDAOLING FRUIT PRODUCTION AND MARKETING PROFESSIONNAL COOPERATIVES平谷区镇罗营镇 ZHENLUOYING,PINGGU DISTRICT,BEIJING 1100GC002 TIANJIN JIZHOU DEVELOPMENT ZONE, WEST IN ZHONGCHANG SOUTH ROAD, NORTH 3 天津蓟州区 JIZHOU,TIANJIN 天津蓟州绿色食品集团有限公司 TIANJIN JIZHOU GREEN FOOD GROUP CO., LTD. 天津市蓟州区开发区中昌南路西、京哈公路北IN JING-HA ROAD 1200GC001 4 河北辛集 XINJI,HEBEI 辛集市裕隆保鲜食品有限责任公司果品包装厂XINJI YULONG FRESHFOOD CO.,LTD. PACKING HOUSE 河北省辛集市南区朝阳路19号 N0.19 CHAOYANG ROAD, SOUTH DISTRICT OF XINJI CITY, HEBEI PROVINCE 1300GC001 5 河北辛集 XINJI,HEBEI 河北天华实业有限公司 HEBEI TIANHUA ENTERPRISE CO.,LTD. 河北省辛集市新垒头村 XINLEITOU VILLAGE,XINJI CITY,HEBEI 1300GC002 6 河北晋州 JINZHOU,HEBEI 河北鲜鲜农产有限公司 HEBEI CICI CO., LTD. 河北省晋州市工业路33号 NO.33 GONGYE ROAD,JINZHOU,HEBEI,CHINA 1300GC004 7 河北晋州 JINZHOU,HEBEI 晋州天洋贸易有限公司 JINZHOU TIANYANG TRADE CO,. LTD. 河北省晋州市通达路 TONGDA ROAD, JINZHOU CITY,HEBEI PROVINCE 1300GC005 8 河北晋州 JINZHOU,HEBEI 河北省晋州市长城经贸有限公司 HEBEI JINZHOU GREAT WALL ECONOMY TRADE CO.,LTD. 河北省晋州市马于开发区 MAYU,JINZHOU,HEBEI,CHINA 1300GC006 9 河北晋州 JINZHOU,HEBEI 石家庄市丰达金润农产品有限公司 SHIJIAZHUANG GOLDEN GLORY AGRICULTURAL CO.,LTD. 晋州市马于镇北辛庄村 BEIXINZHUANG,JINZHOU,HEBEI,CHINA 1300GC007 10 河北赵县 ZHAO COUNTY,HEBEI 河北嘉华农产品有限责任公司 HEBEI JIAHUA -
A New Species of the Genus Takydromus (Squamata, Lacertidae) from Southwestern Guangdong, China
A peer-reviewed open-access journal ZooKeys 871: 119–139 (2019) A new species of Takydromus 119 doi: 10.3897/zookeys.871.35947 RESEARCH ARTICLE http://zookeys.pensoft.net Launched to accelerate biodiversity research A new species of the genus Takydromus (Squamata, Lacertidae) from southwestern Guangdong, China Jian Wang1, Zhi-Tong Lyu1, Chen-Yu Yang1, Yu-Long Li1, Ying-Yong Wang1 1 State Key Laboratory of Biocontrol / The Museum of Biology, School of Life Sciences, Sun Yat-sen University, Guangzhou 510275, China Corresponding author: Ying-Yong Wang ([email protected]) Academic editor: Thomas Ziegler | Received 6 May 2019 | Accepted 31 July2019 | Published 12 August 2019 http://zoobank.org/9C5AE6F4-737C-4E94-A719-AB58CC7002F3 Citation: Wang J, Lyu Z-T, Yang C-Y, Li Y-L, Wang Y-Y (2019) A new species of the genus Takydromus (Squamata, Lacertidae) from southwestern Guangdong, China. ZooKeys 871: 119–139. https://doi.org/10.3897/zookeys.871.35947 Abstract A new species, Takydromus yunkaiensis J. Wang, Lyu, & Y.Y. Wang, sp. nov. is described based on a series of specimens collected from the Yunkaishan Nature Reserve located in the southern Yunkai Mountains, western Guangdong Province, China. The new species is a sister taxon toT. intermedius with a genetic divergence of 8.0–8.5% in the mitochondrial cytochrome b gene, and differs from all known congeners by a combination of the following morphological characters: (1) body size moderate, SVL 37.8–56.0 mm in males, 42.6–60.8 mm in females; (2) dorsal ground color brown; ventral surface -

Ji'an Literati and the Local in Song-Yuan-Ming China
Ji’an Literati and the Local in Song-Yuan-Ming China gerritsen_f1_prelims.indd i 2/6/2007 6:56:53 PM China Studies Published for the Institute for Chinese Studies University of Oxford Editors Glen Dudbridge Frank Pieke VOLUME 13 gerritsen_f1_prelims.indd ii 2/6/2007 6:56:53 PM Ji’an Literati and the Local in Song-Yuan-Ming China By Anne Gerritsen LEIDEN • BOSTON 2007 gerritsen_f1_prelims.indd iii 2/6/2007 6:56:53 PM On the cover : Fragment of a Song dynasty inscription in the Jishui County Museum, Jiangxi province. Photograph by author. This book is printed on acid-free paper. Library of Congress Cataloging-in-Publication Data A C.I.P. record for this book is available from the Library of Congress. ISSN 1570-1344 ISBN 978 90 04 15603 6 © Copyright 2007 by Koninklijke Brill NV, Leiden, The Netherlands. Koninklijke Brill NV incorporates the imprints Brill, Hotei Publishing, IDC Publishers, Martinus Nijhoff Publishers and VSP. All rights reserved. No part of this publication may be reproduced, translated, stored in a retrieval system, or transmitted in any form or by any means, electronic, mechanical, photocopying, recording or otherwise, without prior written permission from the publisher. Authorization to photocopy items for internal or personal use is granted by Koninklijke Brill NV provided that the appropriate fees are paid directly to The Copyright Clearance Center, 222 Rosewood Drive, Suite 910, Danvers, MA 01923, USA. Fees are subject to change. printed in the netherlands gerritsen_f1_prelims.indd iv 2/6/2007 6:56:53 PM To my parents gerritsen_f1_prelims.indd v 2/6/2007 6:56:53 PM gerritsen_f1_prelims.indd vi 2/6/2007 6:56:53 PM CONTENTS List of Maps .............................................................................. -
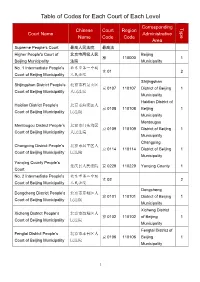
Table of Codes for Each Court of Each Level
Table of Codes for Each Court of Each Level Corresponding Type Chinese Court Region Court Name Administrative Name Code Code Area Supreme People’s Court 最高人民法院 最高法 Higher People's Court of 北京市高级人民 Beijing 京 110000 1 Beijing Municipality 法院 Municipality No. 1 Intermediate People's 北京市第一中级 京 01 2 Court of Beijing Municipality 人民法院 Shijingshan Shijingshan District People’s 北京市石景山区 京 0107 110107 District of Beijing 1 Court of Beijing Municipality 人民法院 Municipality Haidian District of Haidian District People’s 北京市海淀区人 京 0108 110108 Beijing 1 Court of Beijing Municipality 民法院 Municipality Mentougou Mentougou District People’s 北京市门头沟区 京 0109 110109 District of Beijing 1 Court of Beijing Municipality 人民法院 Municipality Changping Changping District People’s 北京市昌平区人 京 0114 110114 District of Beijing 1 Court of Beijing Municipality 民法院 Municipality Yanqing County People’s 延庆县人民法院 京 0229 110229 Yanqing County 1 Court No. 2 Intermediate People's 北京市第二中级 京 02 2 Court of Beijing Municipality 人民法院 Dongcheng Dongcheng District People’s 北京市东城区人 京 0101 110101 District of Beijing 1 Court of Beijing Municipality 民法院 Municipality Xicheng District Xicheng District People’s 北京市西城区人 京 0102 110102 of Beijing 1 Court of Beijing Municipality 民法院 Municipality Fengtai District of Fengtai District People’s 北京市丰台区人 京 0106 110106 Beijing 1 Court of Beijing Municipality 民法院 Municipality 1 Fangshan District Fangshan District People’s 北京市房山区人 京 0111 110111 of Beijing 1 Court of Beijing Municipality 民法院 Municipality Daxing District of Daxing District People’s 北京市大兴区人 京 0115 -

45022-002: Jiangxi Ji'an Sustainable Urban
Social Monitoring Report Project Number: 45022-002 Semi-Annual Report November 2017 PRC: Jiangxi Ji’an Sustainable Urban Transport Project Prepared by Jiangxi Academy of Social Science for the People’s Republic of China and the Asian Development Bank. This social monitoring report is a document of the borrower. The views expressed herein do not necessarily represent those of ADB's Board of Directors, Management, or staff, and may be preliminary in nature. In preparing any country program or strategy, financing any project, or by making any designation of or reference to a particular territory or geographic area in this document, the Asian Development Bank does not intend to make any judgments as to the legal or other status of any territory or area. Asian Development Bank 3216-PRC ADB Loan Ji’an Sustainable Urban Transport Project External Social and Resettlement Monitoring and Evaluation No.1 Report (April 2017 to September 2017) Jiangxi Academy of Social Science November 2017 Executive Abstract According to the ADB’s requirement, the external monitoring of resettlement will be carried out once every six months during the resettlement implementation. The team of EM carried out a monitoring and evaluation on implementation course of LA, HD and resettlement from April to September 2017. The team adopted document method, sampling survey and depth interview method (including interview with affected households and heads of EA.) The EM results show both five roads involving LA and HD. The expropriated land and housing carried out state polices, and met with the standards of resettlement plan approved by ADB. Among these projects, all land were acquired and paid for compensation. -
简报 in February 2016 2016年2月 2016年2月 中国社会福利基金会免费午餐基金管理委员会主办
免费午餐基金 FREE LUNCH FOR CHILDREN BRIEFING简报 IN FEBRUARY 2016 2016年2月 2016年2月 中国社会福利基金会免费午餐基金管理委员会主办 www.mianfeiwucan.org 学校执行汇报 Reports of Registered Schools: 截止2016年2月底 累计开餐学校 517 所 现有开餐学校 437所 现项目受惠人数 143359人 现有用餐人数 104869人 分布于全国23个省市自治区 By the end of February 2016, Free Lunch has found its footprints in 517 schools (currently 437) across 23 provinces, municipalities and autonomous regions with a total of 143,359 beneficiaries and 104,869 currently registered for free lunches. 学校执行详细情况 Details: 2月免费午餐新开餐学校3所, 其中湖南1所,新疆1所,河北1所。 Additional 3 schools are included in the Free Lunch campaign, including 1 in Hunan, 1 in Xinjiang and 1 in Hebei. 学校开餐名单(以拨款时间为准) List of Schools(Grant date prevails): 学校编号 学校名称 微博地址 School No. School Name Weibo Link 2016001 湖南省张家界市桑植县利福塔乡九天洞苗圃学校 http://weibo.com/u/5608665513 Miaopu School of Jiutiandong village, Lifuta Town, Sangzhi County,Zhangjiajie,Hunan Province 2016002 新疆维吾尔族自治区阿克苏地区拜城县赛里木镇英巴格村小学 http://weibo.com/u/5735367760 Primary School of Yingbage Village, Sailimu Town, Baicheng County, Aksu Prefecture, Xinjiang Uygur Autonomous Region 2016003 河北省张家口市赤城县大海陀乡中心小学 http://weibo.com/5784417643 Central Primary School of Dahaituo Town, Chicheng County, Zhangjiakou, Hebei Province 更多学校信息请查看免费午餐官网学校公示页面 For more information about the schools, please view the school page at our official website http://www.mianfeiwucan.org/school/schoolinfo/ 财务数据公示 Financial Data: 2016年2月善款收入: 701万余元,善款支出: 499万余元 善款支出 499万 善款收入 701万 累计总收入 18829万 Donations received: RMB 7.01 million+; Expenditure: RMB 4.99 million+ As of the end of February 2016, Free Lunch for Children has a gross income of RMB 188.29 million. 您可进入免费午餐官网查询捐赠 You can check your donation at our official website: http://www.mianfeiwucan.org/donate/donation/ 项目优秀学校评选表彰名单 Outstanding Schools Name List: 寒假期间,我们对免费午餐的项目执行学校进行了评选表彰活动,感谢学校以辛勤的工作为孩子们带来 温暖安全的午餐。 During the winter holiday, we selected outstanding schools which implemented Free Lunch For Children Program, in recognition of hard work of school staff in providing students warm and safe lunches. -

Download 6.97 MB
Environmental Impact Assessment (Update) Project Number: 45022-002 July 2020 PRC: Jiangxi Ji’an Sustainable Urban Transport Project Prepared by the Ji’an Municipal Government for the People’s Republic of China and the Asian Development Bank. This environmental impact assessment report is a document of the borrower. The views expressed herein do not necessarily represent those of ADB's Board of Directors, Management, or staff, and may be preliminary in nature. In preparing any country program or strategy, financing any project, or by making any designation of or reference to a particular territory or geographic area in this document, the Asian Development Bank does not intend to make any judgments as to the legal or other status of any territory or area. Asian Development Bank CURRENCY EQUIVALENTS (as of 30 June 2020) Currency unit – yuan (CNY) CNY 1.00 = $0. 14 12 $1.00 = CNY 7. 0811 ABBREVIATIONS ADB – Asian Development Bank AQG – air quality guideline As – arsenic B – boron BOD 5 – five-day biochemical oxygen demand BRT – bus rap id transit C&D – construction and demolition Cd – cadmium CN – cyanide CNY – Chinese yuan CO – carbon monoxide CO 2 – carbon dioxide CO 2eq – carbon dioxide equivalent COD – chemical oxyge n dem and CPS – country partnership strategy Cr – chromium Cu – copper DDT – dichloro -diphenyl -trichloroethane DO – dissolved oxygen EA – executing agency EHS – environmental health and safety EIA – environmental impact assessment EIR – en viron mental impact re port EIRF – environme ntal impac t registration form EIT – -

English/Content/Cqu05-06.Pdf
47696 Public Disclosure Authorized MID-TERM EVALUATION OF CHINA’S 11TH 5 YEAR PLAN Public Disclosure Authorized Public Disclosure Authorized Poverty Reduction and Economic Management Unit East Asia and Pacific Region World Bank Public Disclosure Authorized CURRENCY EQUIVALENTS (As of December 18, 2008) Currency = Renminbi Currency Unit = Yuan (CNY) US$1.00 = RMB 6.845 FISCAL YEAR January 1- December 31 WEIGHTS AND MEASURES Metric System ABBREVIATIONS AND ACRONYMS 5YP - Five Year Plan BMI - Basic Medical Insurance BRICs - Brazil, Russia, India, China CHCs - Community Health Centers COD - Chemical Oxygen Demand CPI - Consumer Price Index EFA - Education for All IVDP - Integrated Village Development Program LICs - Low Income Countries MA - Medical Assistance M & E - Monitoring and Evaluation MDG - Millennium Development Goal MEP - Ministry of Environment Protection MICs - Middle Income Countries MOCA - Ministry of Civil Affairs MOF - Ministry of Finance MOLSS/ - Ministry of Labor and Social Security/Ministry of Human Resources (MOHRSS) and Social Security NCH - National Commission on Health NCMS - New Rural Cooperative Medical System NDRC - National Development and Reform Commission OECD - Organization for Economic Co-operation and Development PBOC - People’s Bank of China PPI - Producer Price Index R & D - Research and Development SAT - State Administration of Taxation SO2 - Sulphur Dioxide URBMI - Urban Residents Basic Medical Insurance Vice President: James Adams Country Director: David Dollar Sector Director: Vikram Nehru Task Team Leader: -

World Bank Document
The World Bank Jiangxi Integrated Rural and Urban Water Supply and Wastewater Management Project (P158760) Public Disclosure Authorized Public Disclosure Authorized Combined Project Information Documents / Integrated Safeguards Datasheet (PID/ISDS) Appraisal Stage | Date Prepared/Updated: 15-Jan-2018 | Report No: PIDISDSA21046 Public Disclosure Authorized Public Disclosure Authorized Dec 27, 2017 Page 1 of 26 The World Bank Jiangxi Integrated Rural and Urban Water Supply and Wastewater Management Project (P158760) BASIC INFORMATION OPS_TABLE_BASIC_DATA A. Basic Project Data Country Project ID Project Name Parent Project ID (if any) China P158760 Jiangxi Integrated Rural and Urban Water Supply and Wastewater Management Project Region Estimated Appraisal Date Estimated Board Date Practice Area (Lead) EAST ASIA AND PACIFIC 22-Jan-2018 29-Mar-2018 Water Financing Instrument Borrower(s) Implementing Agency Investment Project Financing PEOPLE'S REPUBLIC OF PIU of Jiangxi Provincial CHINA Water Investment Group Under PMO of Jiangxi Provincial Water Bureau Proposed Development Objective(s) The Project Development Objectives (PDOs) are to increase access and improve operating efficiency of the water supply system, and pilot improved wastewater management services in selected counties in Jiangxi Province. Components Expansion, Rehabilitation, and Modernization of Water Supply System Demonstration of Rural Wastewater Management Services Public Engagement and Project Management Financing (in USD Million) Finance OLD Financing Source Amount Borrower 164.74 International Bank for Reconstruction and Development 200.00 Total Project Cost 364.74 Environmental Assessment Category B - Partial Assessment Decision The review did authorize the preparation to continue Dec 27, 2017 Page 2 of 26 The World Bank Jiangxi Integrated Rural and Urban Water Supply and Wastewater Management Project (P158760) Other Decision (as needed) B.