Risk Assessment and Risk Mitigation Review(S)
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Therapeutic Use of a Selective S1P1 Receptor Modulator Ponesimod in Autoimmune Diabetes
Therapeutic Use of a Selective S1P1 Receptor Modulator Ponesimod in Autoimmune Diabetes Sylvaine You1,2, Luca Piali3, Chantal Kuhn1,2, Beat Steiner3, Virginia Sauvaget1,2, Fabrice Valette1,2, Martine Clozel3, Jean-Franc¸ois Bach1,2, Lucienne Chatenoud1,2* 1 Universite´ Paris Descartes, Sorbonne Paris Cite´, Paris, France, 2 Institut National de la Sante´ et de la Recherche Me´dicale, Unite´ 1013, Paris, France, 3 Actelion Pharmaceuticals Ltd, Allschwil, Switzerland Abstract In the present study, we investigated the therapeutic potential of a selective S1P1 receptor modulator, ponesimod, to protect and reverse autoimmune diabetes in non-obese diabetic (NOD) mice. Ponesimod was administered orally to NOD mice starting at 6, 10, 13 and 16 weeks of age up to 35 weeks of age or to NOD mice showing recent onset diabetes. Peripheral blood and spleen B and T cell counts were significantly reduced after ponesimod administration. In pancreatic lymph nodes, B lymphocytes were increased and expressed a transitional 1-like phenotype. Chronic oral ponesimod treatment efficiently prevented autoimmune diabetes in 6, 10 and 16 week-old pre-diabetic NOD mice. Treatment withdrawal led to synchronized disease relapse. Ponesimod did not inhibit the differentiation of autoreactive T cells as assessed by adoptive transfer of lymphocytes from treated disease-free NOD mice. In addition, it did not affect the migration, proliferation and activation of transgenic BDC2.5 cells into the target tissue. However, ponesimod inhibited spreading of the T cell responses to islet-specific glucose-6-phosphatase catalytic subunit-related protein (IGRP). Treatment of diabetic NOD mice with ponesimod induced disease remission. However, here again, upon treatment cessation, the disease rapidly recurred. -

Sphingosine-1-Phosphate: Its Pharmacological Regulation and the Treatment of Multiple Sclerosis: a Review Article
biomedicines Review Sphingosine-1-Phosphate: Its Pharmacological Regulation and the Treatment of Multiple Sclerosis: A Review Article Stanley Cohan, Elisabeth Lucassen, Kyle Smoot, Justine Brink and Chiayi Chen * Providence Multiple Sclerosis Center, Providence Brain and Spine Institute, Providence St, Vincent Medical Center, Portland, OR 97225, USA; [email protected] (S.C.); [email protected] (E.L.); [email protected] (K.S.); [email protected] (J.B.) * Correspondence: [email protected]; Tel.: +1-503-216-1012 Received: 27 May 2020; Accepted: 15 July 2020; Published: 18 July 2020 Abstract: Sphingosine-1-phosphate (S1P), via its G-protein-coupled receptors, is a signaling molecule with important regulatory properties on numerous, widely varied cell types. Five S1P receptors (S1PR1-5) have been identified, each with effects determined by their unique G-protein-driven downstream pathways. The discovery that lymphocyte egress from peripheral lymphoid organs is promoted by S1P via S1PR-1 stimulation led to the development of pharmacological agents which are S1PR antagonists. These agents promote lymphocyte sequestration and reduce lymphocyte-driven inflammatory damage of the central nervous system (CNS) in animal models, encouraging their examination of efficacy in the treatment of multiple sclerosis (MS). Preclinical research has also demonstrated direct protective effects of S1PR antagonists within the CNS, by modulation of S1PRs, particularly S1PR-1 and S1PR-5, and possibly S1PR-2, independent of effects upon lymphocytes. Three of these agents, fingolimod, siponimod and ozanimod have been approved, and ponesimod has been submitted for regulatory approval. In patients with MS, these agents reduce relapse risk, sustained disability progression, magnetic resonance imaging markers of disease activity, and whole brain and/or cortical and deep gray matter atrophy. -

Oral MS Disease-Modifying Therapies C21142-A
Drug and Biologic Coverage Criteria Effective Date: 05/01/2019 Last P&T Approval/Version: 07/28/2021 Next Review Due By: 08/2022 Policy Number: C21142-A Oral MS Disease-Modifying Therapies PRODUCTS AFFECTED Mayzent (siponimod), Aubagio (teriflunomide), Gilenya (fingolimod), Mavenclad (cladribine), Tecfidera (dimethyl fumarate), Vumerity (diroximel fumarate), Bafiertam (monomethyl fumarate),dimethyl fumarate, Zeposia (ozanimod), Ponvory (ponesimod) COVERAGE POLICY Coverage for services, procedures, medical devices, and drugs are dependent upon benefit eligibility as outlined in the member's specific benefit plan. This Coverage Guideline must be read in its entirety to determine coverage eligibility, if any. This Coverage Guideline provides information related to coverage determinations only and does not imply that a service or treatment is clinically appropriate or inappropriate. The provider and the member are responsible for all decisions regarding the appropriateness of care. Providers should provide Molina Healthcare complete medical rationale when requesting any exceptions to these guidelines Documentation Requirements: Molina Healthcare reserves the right to require that additional documentation be made available as part of its coverage determination; quality improvement; and fraud; waste and abuse prevention processes. Documentation required may include, but is not limited to, patient records, test results and credentials of the provider ordering or performing a drug or service. Molina Healthcare may deny reimbursement or take additional appropriate action if the documentation provided does not support the initial determination that the drugs or services were medically necessary, not investigational or experimental, and otherwise within the scope of benefits afforded to the member, and/or the documentation demonstrates a pattern of billing or other practice that is inappropriate or excessive DIAGNOSIS: Multiple Sclerosis REQUIRED MEDICAL INFORMATION: A. -

Multiple Sclerosis
© Copyright 2012 Oregon State University. All Rights Reserved Drug Use Research & Management Program Oregon State University, 500 Summer Street NE, E35 Salem, Oregon 97301-1079 Phone 503-947-5220 | Fax 503-947-2596 Drug Class Update with New Drug Evaluations: Multiple Sclerosis Date of Review: June 2021 Date of Last Review: August 2020 Dates of Literature Search: 02/03/2020 – 03/01/2021 Generic Name: Ofatumumab Ponesimod Brand Name (Manufacturer): Kesimpta (Novartis) Ponvory (Janssen) Dossier Received: yes, for ofatumumab Current Status of PDL Class: See Appendix 1. Purpose for Class Update: Evidence for the comparative effectiveness of disease modifying drugs (DMDs) for multiple sclerosis (MS) was last reviewed by the Oregon Pharmacy & Therapeutic Committee (P&T) in August 2020 as summarized in a Drug Effectiveness Review Project (DERP) report. This review examines new comparative evidence of DMDs for MS published since 2020 and summarizes the evidence for 2 new DMDs, ofatumumab and ponesimod, approved to treat relapsing forms of MS. Research Questions: 1. What is the comparative effectiveness and efficacy of DMDs for MS? 2. Do DMDs for MS differ in harms? 3. Are there subgroups of patients with MS based on demographics (age, racial or ethnic groups, and gender), socioeconomic status, concomitant medications, severity of disease, or co-morbidities for which one DMD is more effective or associated with fewer adverse events? 4. What is the evidence for efficacy and safety for use of ofatumumab in relapsing MS? 5. What is the evidence for efficacy and safety of ponesimod in relapsing MS ? Author: Deanna Moretz, PharmD, BCPS Conclusions: Multiple Sclerosis Disease-Modifying Drugs No new evidence of comparative efficacy or effectiveness of DMDs approved to treat MS has been published since the last MS class review. -

Ponesimod (Ponvory) Reference Number: ERX.SPA.437 Effective Date: 06.01.21 Last Review Date: 05.21 Line of Business: Commercial, Medicaid Revision Log
Clinical Policy: Ponesimod (Ponvory) Reference Number: ERX.SPA.437 Effective Date: 06.01.21 Last Review Date: 05.21 Line of Business: Commercial, Medicaid Revision Log See Important Reminder at the end of this policy for important regulatory and legal information. Description Ponesimod (Ponvory™) is a sphingosine 1-phosphate receptor modulator. FDA Approved Indication(s) Ponvory is indicated for the treatment of relapsing forms of multiple sclerosis (MS), to include clinically isolated syndrome, relapsing-remitting disease, and active secondary progressive disease, in adults. Policy/Criteria Provider must submit documentation (such as office chart notes, lab results or other clinical information) supporting that member has met all approval criteria. Health plan approved formularies should be reviewed for all coverage determinations. Requirements to use preferred alternative agents apply only when such requirements align with the health plan approved formulary. It is the policy of health plans affiliated with Envolve Pharmacy Solutions™ that Ponvory is medically necessary when the following criteria are met: I. Initial Approval Criteria A. Multiple Sclerosis (must meet all): 1. Diagnosis of one of the following (a, b, or c): a. Clinically isolated syndrome, and member is contraindicated to all, or has experienced clinically significant adverse effects to two, of the following: Aubagio®, glatiramer (Copaxone®, Glatopa®), an interferon-beta agent (Betaseron® or Rebif®); b. Relapsing-remitting MS, and failure of two of the following, unless clinically significant adverse effects are experienced or all are contraindicated: Aubagio®, dimethyl fumarate, Gilenya®, glatiramer (Copaxone®, Glatopa®), an interferon-beta agent (Betaseron® or Rebif®), Kesimpta®, Mayzent®, Ocrevus®, Tysabri®, Vumerity®, Zeposia®;* *Prior authorization is required for all disease modifying therapies for MS. -

Rxoutlook® 4Th Quarter 2020
® RxOutlook 4th Quarter 2020 optum.com/optumrx a RxOutlook 4th Quarter 2020 While COVID-19 vaccines draw most attention, multiple “firsts” are expected from the pipeline in 1Q:2021 Great attention is being given to pipeline drugs that are being rapidly developed for the treatment or prevention of SARS- CoV-19 (COVID-19) infection, particularly two vaccines that are likely to receive emergency use authorization (EUA) from the Food and Drug Administration (FDA) in the near future. Earlier this year, FDA issued a Guidance for Industry that indicated the FDA expected any vaccine for COVID-19 to have at least 50% efficacy in preventing COVID-19. In November, two manufacturers, Pfizer and Moderna, released top-line results from interim analyses of their investigational COVID-19 vaccines. Pfizer stated their vaccine, BNT162b2 had demonstrated > 90% efficacy. Several days later, Moderna stated their vaccine, mRNA-1273, had demonstrated 94% efficacy. Many unknowns still exist, such as the durability of response, vaccine performance in vulnerable sub-populations, safety, and tolerability in the short and long term. Considering the first U.S. case of COVID-19 was detected less than 12 months ago, the fact that two vaccines have far exceeded the FDA’s guidance and are poised to earn EUA clearance, is remarkable. If the final data indicates a positive risk vs. benefit profile and supports final FDA clearance, there may be lessons from this accelerated development timeline that could be applied to the larger drug development pipeline in the future. Meanwhile, drug development in other areas continues. In this edition of RxOutlook, we highlight 12 key pipeline drugs with potential to launch by the end of the first quarter of 2021. -

Benefit–Risk Profile of Sphingosine-1-Phosphate
Drugs (2017) 77:1755–1768 DOI 10.1007/s40265-017-0814-1 REVIEW ARTICLE Benefit–Risk Profile of Sphingosine-1-Phosphate Receptor Modulators in Relapsing and Secondary Progressive Multiple Sclerosis 1 2,3 4 5 Giancarlo Comi • Hans-Peter Hartung • Rajesh Bakshi • Ian M. Williams • Heinz Wiendl6 Published online: 13 September 2017 Ó The Author(s) 2017. This article is an open access publication Abstract Since the approval of fingolimod, several selec- studies demonstrated sustained efficacy and raised no new tive sphingosine-1-phosphate receptor modulators have safety concerns, with no increases in macular edema, entered clinical development for multiple sclerosis. How- infection, or malignancy rates. Switch studies identified no ever, side effects can occur with sphingosine-1-phosphate safety concerns and greater patient satisfaction and per- receptor modulators. By considering short-term data across sistence with fingolimod when switching from the drug class and longer term fingolimod data, we aim to injectable therapies with no washout period. Better out- highlight the potential of sphingosine-1-phosphate receptor comes were seen with short than with long washouts when modulators in multiple sclerosis, while offering reassur- switching from natalizumab. The specific immunomodu- ance that their benefit–risk profiles are suitable for long- latory effects of sphingosine-1-phosphate receptor modu- term therapy. Short-term fingolimod studies demonstrated lators are consistent with the low observed rates of long- the efficacy of this drug class, showed that cardiac events term, drug-related adverse effects with fingolimod. Short- upon first-dose administration are transient and manage- term data for selective sphingosine-1-phosphate receptor able, and showed that serious adverse events are rare. -

Biologics for Autoimmune Conditions
© Copyright 2021 Oregon State University. All Rights Reserved Drug Use Research & Management Program Oregon State University, 500 Summer Street NE, E35 Salem, Oregon 97301-1079 Phone 503-947-5220 | Fax 503-947-2596 Drug Class Update: Targeted Immune Modulators for Autoimmune Diseases Date of Review: October 2021 Date of Last Review: October 2020 Dates of Literature Search: 01/01/2020 – 06/09/2021 Current Status of PDL Class: See Appendix 1. Purpose for Class Update: Review new comparative evidence between targeted immune modulators (TIMs) approved by the Food and Drug Administration (FDA) to manage autoimmune conditions. Research Questions: 1. Is there new comparative evidence that TIMs differ in efficacy or effectiveness for alleviating symptoms and stabilizing disease in patients with rheumatoid arthritis (RA), ankylosing spondylitis (AS), psoriatic arthritis (PsA), plaque psoriasis (PsO), Crohn’s disease (CD), or ulcerative colitis (UC)? 2. Is there new comparative evidence that TIMs differ in harms when used to treat patients with autoimmune conditions? 3. Are there specific subpopulations for which one TIM is better tolerated or more effective than other available TIMs when used for autoimmune conditions? Conclusions: Since the last class update, 6 high-quality systematic reviews and 5 high-quality guidelines were published. Systematic Reviews In April 2020 the American Gastroenterological Association (AGA) sponsored a systematic review focused on the efficacy and safety of disease-modifying anti-rheumatic drugs (DMARDs) approved -

Ponesimod MBMA ARR Abstract for ECF 2020 Confidential Title
Ponesimod_MBMA_ARR Abstract for ECF 2020 Confidential Title: Comparative Efficacy of Relapsing Multiple Sclerosis Therapies: A Model-Based Meta-Analysis for Annualized Relapse Rate Short title: Comparative Efficacy of Relapsing MS Therapies Authors: 1M. Zierhut, 2H. Kracker, 2T. Scherz, 3A. Keenan, 2B. Hennessy Affiliations: 1Janssen Research & Development, San Diego, CA, United States of America, 2Actelion Pharmaceuticals Ltd, Part of Janssen Pharmaceutical Companies, Allschwil, Switzerland, 3Janssen Research & Development, LLC, Spring House, United States of America Corresponding Author: Alexander Keenan Global Market Access Leader, Schizophrenia and Neurodegeneration Janssen Research & Development, LLC Spring House, Pennsylvania E-mail: [email protected] Funding: This work was sponsored by Janssen Research & Development, LLC. Previous submission: A/ECTRIMS 2020, September 9-12, 2020, Washington DC Ponesimod_MBMA_ARR Abstract for ECF 2020 Confidential Abstract (300/300 words) Background: Multiple sclerosis (MS), an inflammatory autoimmune disorder, is responsible for progressive neurological disability among adults. Ponesimod is a selective sphingosine-1- phosphate (S1P) receptor 1 immunomodulator under development for relapsing multiple sclerosis (RMS). Objectives: To assess effects of ponesimod on the annualized relapse rate (ARR) relative to other disease-modifying therapies (DMTs) used for treatment of RMS. Methods: A literature review was performed, returning 154 clinical trials with 58 MS treatments. Only randomized controlled trials (RCTs) with >30 patients receiving monotherapy for RMS for ≥48 weeks were included. Mean ARR for each treatment was modeled and rate ratios (RRs) between treatments were assumed constant from 48-156 weeks. Multiple variables were explored relative treatment effect modifiers: percent of patients with remitting RMS, trial start year, mean disease duration, percent of patients with history of DMT use in<2 years (pDMT), mean relapses, mean age, and mean baseline EDSS score. -

Ponvory (Ponesimod) Annual Review Date: 05/20/2021
Policy: Ponvory (ponesimod) Annual Review Date: 05/20/2021 Last Revised Date: 05/20/2021 OVERVIEW Ponvory, a sphingosine 1-phosphate receptor modulator, is indicated for the treatment of patients with relapsing forms of multiple sclerosis (MS), to include clinically isolated syndrome, relapsing remitting disease, and active secondary progressive disease in adults. POLICY STATEMENT This policy involves the use of Ponvory. Prior authorization is recommended for pharmacy benefit coverage of Ponvory. Approval is recommended for those who meet the conditions of coverage in the Criteria and Initial/Extended Approval for the diagnosis provided. Conditions Not Recommended for Approval are listed following the recommended authorization criteria. Requests for uses not listed in this policy will be reviewed for evidence of efficacy and for medical necessity on a case-by-case basis. Because of the specialized skills required for evaluation and diagnosis of patients treated with Ponvory as well as the monitoring required for adverse events and long-term efficacy, initial approval requires Ponvory be prescribed by or in consultation with a physician who specializes in the condition being treated. All approvals for initial therapy are provided for the initial approval duration noted below; if reauthorization is allowed, a response to therapy is required for continuation of therapy unless otherwise noted below. RECOMMENDED AUTHORIZATION CRITERIA Coverage of Ponvory is recommended in those who meet the following criteria: 1. Multiple Sclerosis Criteria. Patient must meet the following criteria A) Patient has a relapsing form of multiple sclerosis; AND Note: Examples of relapsing forms of multiple sclerosis include clinically isolated syndrome, relapsing remitting disease, and active secondary progressive disease. -
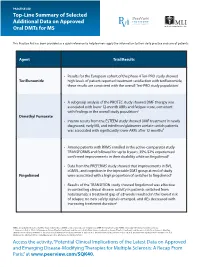
Top-Line Summary of Selected Additional Data on Approved Oral Dmts for MS
PRACTICE AID Top-Line Summary of Selected Additional Data on Approved Oral DMTs for MS This Practice Aid has been provided as a quick reference to help learners apply the information to their daily practice and care of patients. Agent Trial/Results • Results for the European cohort of the phase 4 Teri-PRO study showed Teriflunomide high levels of patient-reported treatment satisfaction with teriflunomide; these results are consistent with the overall Teri-PRO study population1 • A subgroup analysis of the PROTEC study showed DMF therapy was associated with lower 12-month ARRs and fatigue score, consistent with findings in the overall study population2 Dimethyl Fumarate • Interim results from the ESTEEM study showed DMF treatment in newly diagnosed, early MS, and interferon/glatiramer acetate switch patients was associated with significantly lower ARRs after 12 months3 • Among patients with RRMS enrolled in the active-comparator study TRANSFORMS and followed for up to 8 years, 35%-52% experienced confirmed improvements in their disability while on fingolimod4 • Data from the PREFERMS study showed that improvements in BVL, cGMVL, and cognition in the injectable DMT group at end of study Fingolimod were associated with a high proportion of switches to fingolimod5 • Results of the TRANSITION study showed fingolimod was effective in controlling clinical disease activity in patients switched from natalizumab; a treatment gap of ≤8 weeks resulted in the lowest risk of relapse; no new safety signals emerged, and AEs decreased with increasing treatment duration6 ARRs: annualized relapse rates; BVL: brain volume loss; cGMVL: cortical gray matter volume loss; DMF: dimethyl fumarate; RRMS: relapsing-remitting multiple sclerosis. -

Celgene Bets Big on Scripps-Originated Autoimmunity Candidate
NEWS & ANALYSIS BIOBUSINESS BRIEFS DEAL WATCH Celgene bets big on Scripps-originated autoimmunity candidate In its third major acquisition in 3 months, relapsing–remitting multiple sclerosis — Celgene is buying the biotech company fingolimod (Novartis) — and by two others Receptos for US$7.2 billion. In doing so, that are in Phase III trials, siponimod Celgene adds two clinical-stage drug (Novartis) and ponesimod (Actelion). candidates for treating autoimmune diseases to In Europe, fingolimod is only approved for its portfolio, including one — ozanimod — that patients with active disease for whom originated from the US National Institutes of interferon‑β and glatiramer acetate are not Health (NIH) Molecular Libraries Program (MLP). effective. Safety issues with fingolimod — “This is a very rare example of a compound that most notably, fatal infections with varicella Ryan McVay/Photodisc was discovered in academia and has made it zoster virus and decreases in heart rate, into Phase III,” says Hugh Rosen, a professor both of which were observed in clinical trials at The Scripps Research Institute (TSRI) in San — mean there is room for improvement Diego, USA, who co‑discovered ozanimod. among S1PR-targeting compounds. “It looks In 2009, Receptos licensed a series of to me like the cardiac side effects are a bit compounds identified in the laboratories of less with ozanimod,” says Bernhard Hemmer, Rosen and his colleague, Edward Roberts, a professor at Technische Universität from TSRI. The researchers had screened München, Munich, Germany, and an expert the NIH MLP collection to identify allosteric in multiple sclerosis. However, he cautions, agonists of the sphingosine-1-phosphate “one has to see how the drug performs in receptor 1 (S1PR1).