Functional Residues in Proteins
Total Page:16
File Type:pdf, Size:1020Kb

Load more
Recommended publications
-

Review Article Functional Subunits of Eukaryotic Chaperonin CCT/Tric in Protein Folding
SAGE-Hindawi Access to Research Journal of Amino Acids Volume 2011, Article ID 843206, 16 pages doi:10.4061/2011/843206 Review Article Functional Subunits of Eukaryotic Chaperonin CCT/TRiC in Protein Folding M. Anaul Kabir,1 Wasim Uddin,1 Aswathy Narayanan,1 Praveen Kumar Reddy,1 M. Aman Jairajpuri,2 Fred Sherman,3 and Zulfiqar Ahmad4 1 Molecular Genetics Laboratory, School of Biotechnology, National Institute of Technology Calicut, Kerala 673601, India 2 Department of Biosciences, Jamia Millia Islamia, Jamia Nagar, New Delhi 110025, India 3 Department of Biochemistry and Biophysics, University of Rochester Medical Center, NY 14642, USA 4 Department of Biology, Alabama A&M University, Normal, AL 35762, USA Correspondence should be addressed to M. Anaul Kabir, [email protected] Received 15 February 2011; Accepted 5 April 2011 Academic Editor: Shandar Ahmad Copyright © 2011 M. Anaul Kabir et al. This is an open access article distributed under the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited. Molecular chaperones are a class of proteins responsible for proper folding of a large number of polypeptides in both prokaryotic and eukaryotic cells. Newly synthesized polypeptides are prone to nonspecific interactions, and many of them make toxic aggregates in absence of chaperones. The eukaryotic chaperonin CCT is a large, multisubunit, cylindrical structure having two identical rings stacked back to back. Each ring is composed of eight different but similar subunits and each subunit has three distinct domains. CCT assists folding of actin, tubulin, and numerous other cellular proteins in an ATP-dependent manner. -

A-Ailable At
International Journal of Current Advanced Research ISSN: O: 2319-6475, ISSN: P: 2319 – 6505, Impact Factor: SJIF: 5.438 Available Online at www.journalijcar.org Volume 6; Issue 2; February 2017; Page No. 2131-2138 Research Article A COMPARATIVE ANALYSIS OF THERMOPHILIC AND PSYCHROPHILIC METALLOPEPTIDASES: INVOLVEMENT OF BIOINFORMATIC APPROACH Anuprabha1., AbhigyanNath2 and Radha Chaube3* 1,2 3Bioinformatic Division, MahilaMahavidyalya, Banaras Hindu University, Varanasi-221005, Uttar Pradesh, India ARTICLEDepartment INFO of Zoology, Institute of Science,ABSTRACT Banaras Hindu University, Varanasi-221005, Uttar Pradesh, India The thermolysin family of enzymes is classified as the M4 family of metallopeptidases. M4 Article History: family comprises numerous zinc-dependent metallopeptidases that hydrolyze peptide Received 9th November, 2016 bonds. Zinc containing peptidases are widely distributed in nature and have important roles Received in revised form 12thDecember, 2016 in many physiological processes. M4 family comprises numerous zinc-dependent Accepted 5th January, 2017 metallopeptidases that hydrolyse peptide bonds. Metallopeptidases were studied for the Published online 28th February, 2017 reason of their great relevance to biology, medicine and biotechnology. In the present study, detailed comparative analyses of both thermophilic and psychrophilic metallopeptidases were performed. The comparative analysis of both thermophilic and Key words: psychrophilic metallopeptidases was performed by taking sequence parameters such as Thermophilic, Psychrophilic, amino acid composition, amino acid property group composition, physico-chemical Metallopeptidases, amino acid residues, properties, secondary structure content. From comparison between the two groups, it was analysis found that Gln, Asn, Ser, Thr, and His are significantly lower in thermophilic metallopeptidase. Positively charged residues (Lys, Arg and Glu) are more significant in thermophilic metallopeptidases than in psychrophilic metallopeptidases. -

(12) United States Patent (10) Patent No.: US 6,395,889 B1 Robison (45) Date of Patent: May 28, 2002
USOO6395889B1 (12) United States Patent (10) Patent No.: US 6,395,889 B1 Robison (45) Date of Patent: May 28, 2002 (54) NUCLEIC ACID MOLECULES ENCODING WO WO-98/56804 A1 * 12/1998 ........... CO7H/21/02 HUMAN PROTEASE HOMOLOGS WO WO-99/0785.0 A1 * 2/1999 ... C12N/15/12 WO WO-99/37660 A1 * 7/1999 ........... CO7H/21/04 (75) Inventor: fish E. Robison, Wilmington, MA OTHER PUBLICATIONS Vazquez, F., et al., 1999, “METH-1, a human ortholog of (73) Assignee: Millennium Pharmaceuticals, Inc., ADAMTS-1, and METH-2 are members of a new family of Cambridge, MA (US) proteins with angio-inhibitory activity', The Journal of c: - 0 Biological Chemistry, vol. 274, No. 33, pp. 23349–23357.* (*) Notice: Subject to any disclaimer, the term of this Descriptors of Protease Classes in Prosite and Pfam Data patent is extended or adjusted under 35 bases. U.S.C. 154(b) by 0 days. * cited by examiner (21) Appl. No.: 09/392, 184 Primary Examiner Ponnathapu Achutamurthy (22) Filed: Sep. 9, 1999 ASSistant Examiner William W. Moore (51) Int. Cl." C12N 15/57; C12N 15/12; (74) Attorney, Agent, or Firm-Alston & Bird LLP C12N 9/64; C12N 15/79 (57) ABSTRACT (52) U.S. Cl. .................... 536/23.2; 536/23.5; 435/69.1; 435/252.3; 435/320.1 The invention relates to polynucleotides encoding newly (58) Field of Search ............................... 536,232,235. identified protease homologs. The invention also relates to 435/6, 226, 69.1, 252.3 the proteases. The invention further relates to methods using s s s/ - - -us the protease polypeptides and polynucleotides as a target for (56) References Cited diagnosis and treatment in protease-mediated disorders. -

The Extracellular Proteases Produced by Vibrio Parahaemolyticus
World Journal of Microbiology and Biotechnology (2018) 34:68 https://doi.org/10.1007/s11274-018-2453-4 REVIEW The extracellular proteases produced by Vibrio parahaemolyticus George Osei‑Adjei1 · Xinxiang Huang1 · Yiquan Zhang1 Received: 20 February 2018 / Accepted: 8 May 2018 © Springer Science+Business Media B.V., part of Springer Nature 2018 Abstract Vibrio parahaemolyticus, a Gram-negative bacterium, inhabits marine and estuarine environments and it is a major pathogen responsible globally for most cases of seafood-associated gastroenteritis in humans and acute hepatopancreatic necrosis syn- drome in shrimps. There has been a dramatic worldwide increase in V. parahaemolyticus infections over the last two decades. The pathogenicity of V. parahaemolyticus has been linked to the expression of different kinds of virulence factors including extracellular proteases, such as metalloproteases and serine proteases. V. parahaemolyticus expresses the metalloproteases; PrtV, VppC, VPM and the serine proteases; VPP1/Protease A, VpSP37, PrtA. Extracellular proteases have been identified as potential virulence factors which directly digest many kinds of host proteins or indirectly are involved in the processing of other toxic protein factors. This review summarizes findings on the metalloproteases and serine proteases produced by V. parahaemolyticus and their roles in infections. Identifying the role of V. parahaemolyticus virulence-associated extracellular proteases deepens our understanding of diseases caused by this bacterium. Keywords Extracellular protease · Metalloprotease · Serine protease · Vibrio parahaemolyticus · Virulence Introduction incubation period of V. parahaemolyticus is usually 12–24 h post-infection with infection usually occurring in the hot The Gram-negative bacterium, Vibrio parahaemolyticus, summer months because the bacteria is found free swim- lives in marine and estuarine environments and it is a major ming in coastal water or attached to fish and shellfish. -

Serine Proteases with Altered Sensitivity to Activity-Modulating
(19) & (11) EP 2 045 321 A2 (12) EUROPEAN PATENT APPLICATION (43) Date of publication: (51) Int Cl.: 08.04.2009 Bulletin 2009/15 C12N 9/00 (2006.01) C12N 15/00 (2006.01) C12Q 1/37 (2006.01) (21) Application number: 09150549.5 (22) Date of filing: 26.05.2006 (84) Designated Contracting States: • Haupts, Ulrich AT BE BG CH CY CZ DE DK EE ES FI FR GB GR 51519 Odenthal (DE) HU IE IS IT LI LT LU LV MC NL PL PT RO SE SI • Coco, Wayne SK TR 50737 Köln (DE) •Tebbe, Jan (30) Priority: 27.05.2005 EP 05104543 50733 Köln (DE) • Votsmeier, Christian (62) Document number(s) of the earlier application(s) in 50259 Pulheim (DE) accordance with Art. 76 EPC: • Scheidig, Andreas 06763303.2 / 1 883 696 50823 Köln (DE) (71) Applicant: Direvo Biotech AG (74) Representative: von Kreisler Selting Werner 50829 Köln (DE) Patentanwälte P.O. Box 10 22 41 (72) Inventors: 50462 Köln (DE) • Koltermann, André 82057 Icking (DE) Remarks: • Kettling, Ulrich This application was filed on 14-01-2009 as a 81477 München (DE) divisional application to the application mentioned under INID code 62. (54) Serine proteases with altered sensitivity to activity-modulating substances (57) The present invention provides variants of ser- screening of the library in the presence of one or several ine proteases of the S1 class with altered sensitivity to activity-modulating substances, selection of variants with one or more activity-modulating substances. A method altered sensitivity to one or several activity-modulating for the generation of such proteases is disclosed, com- substances and isolation of those polynucleotide se- prising the provision of a protease library encoding poly- quences that encode for the selected variants. -

Proteolytic Enzymes in Grass Pollen and Their Relationship to Allergenic Proteins
Proteolytic Enzymes in Grass Pollen and their Relationship to Allergenic Proteins By Rohit G. Saldanha A thesis submitted in fulfilment of the requirements for the degree of Masters by Research Faculty of Medicine The University of New South Wales March 2005 TABLE OF CONTENTS TABLE OF CONTENTS 1 LIST OF FIGURES 6 LIST OF TABLES 8 LIST OF TABLES 8 ABBREVIATIONS 8 ACKNOWLEDGEMENTS 11 PUBLISHED WORK FROM THIS THESIS 12 ABSTRACT 13 1. ASTHMA AND SENSITISATION IN ALLERGIC DISEASES 14 1.1 Defining Asthma and its Clinical Presentation 14 1.2 Inflammatory Responses in Asthma 15 1.2.1 The Early Phase Response 15 1.2.2 The Late Phase Reaction 16 1.3 Effects of Airway Inflammation 16 1.3.1 Respiratory Epithelium 16 1.3.2 Airway Remodelling 17 1.4 Classification of Asthma 18 1.4.1 Extrinsic Asthma 19 1.4.2 Intrinsic Asthma 19 1.5 Prevalence of Asthma 20 1.6 Immunological Sensitisation 22 1.7 Antigen Presentation and development of T cell Responses. 22 1.8 Factors Influencing T cell Activation Responses 25 1.8.1 Co-Stimulatory Interactions 25 1.8.2 Cognate Cellular Interactions 26 1.8.3 Soluble Pro-inflammatory Factors 26 1.9 Intracellular Signalling Mechanisms Regulating T cell Differentiation 30 2 POLLEN ALLERGENS AND THEIR RELATIONSHIP TO PROTEOLYTIC ENZYMES 33 1 2.1 The Role of Pollen Allergens in Asthma 33 2.2 Environmental Factors influencing Pollen Exposure 33 2.3 Classification of Pollen Sources 35 2.3.1 Taxonomy of Pollen Sources 35 2.3.2 Cross-Reactivity between different Pollen Allergens 40 2.4 Classification of Pollen Allergens 41 2.4.1 -
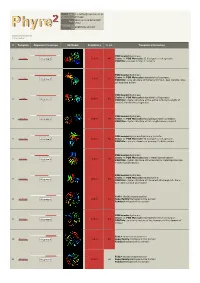
Phyre 2 Results for P22894
Email [email protected] Description P22894 Wed Jun 6 09:54:50 BST Date 2012 Unique Job eba6f50a2e10c91f ID Detailed template information # Template Alignment Coverage 3D Model Confidence % i.d. Template Information PDB header:hydrolase 1 c1gxdA_ Alignment 100.0 46 Chain: A: PDB Molecule:72 kda type iv collagenase; PDBTitle: prommp-2/timp-2 complex PDB header:hydrolase Chain: A: PDB Molecule:interstitial collagenase; 2 c1su3A_ 100.0 61 Alignment PDBTitle: x-ray structure of human prommp-1: new insights into2 collagenase action PDB header:hydrolase Chain: B: PDB Molecule:interstitial collagenase; 3 c2cltB_ 100.0 61 Alignment PDBTitle: crystal structure of the active form (full-length) of human2 fibroblast collagenase. PDB header:hydrolase 4 c3ba0A_ Alignment 100.0 49 Chain: A: PDB Molecule:macrophage metalloelastase; PDBTitle: crystal structure of full-length human mmp-12 PDB header:hydrolase/hydrolase inhibitor 5 c1eakA_ Alignment 100.0 52 Chain: A: PDB Molecule:72 kda type iv collagenase; PDBTitle: catalytic domain of prommp-2 e404q mutant PDB header:hydrolase Chain: A: PDB Molecule:matrix metalloproteinase-9; 6 c1l6jA_ 100.0 56 Alignment PDBTitle: crystal structure of human matrix metalloproteinase mmp92 (gelatinase b). PDB header:hydrolase Chain: A: PDB Molecule:stromelysin-1; 7 c1slmA_ 100.0 54 Alignment PDBTitle: crystal structure of fibroblast stromelysin-1: the c- truncated human2 proenzyme Fold:4-bladed beta-propeller 8 d1fbla1 Alignment 100.0 57 Superfamily:Hemopexin-like domain Family:Hemopexin-like domain PDB header:hydrolase -

Structure, Hormonal Regulation and Chromosomal Location of Genes Encoding Barley (1-+A)-B-Xylan Endohydrolases
l;),, (!. s,9 47 STRUCTURE, HORMONAL RB,GULA CHROMOSOMAL LOCATION OF GENES ENCODING BARLEY (1+4)- p-XYLAI\ ENDOHYDROLASES by MITALI BANIK, B.Sc. (Hons.), M.Sc. A thesis submitted in fulfillment of the requirement for the degree of Doctor of Philosophy Department of Plant Science, Division of Agricultural and Natural Resource Sciences, TheUniversity of Adelaide, Waite Campus, Glen Osmond,5064 Australia October, 1996 1l STATEMENT OF AUTHORSHIP Except where reference is made in the text of the thesis, this thesis contains no material published elsewhere or extracted in whole or in part from a thesis presented by me for another degree or diploma. No other person's work has been used without due acknowledgment in the main text of the thesis. This thesis has not been submitted for the award of any other degree or diploma in any other tertiary institution. MITALI BANIK October, 1996 o lll ACKNOWLBDGMENTS I would like to express my deepest sense of gratitude and indebtedness to my supervisor, Professor Geoffrey B. Fincher, Head of Department, Plant Science, University of Adelaide for his constant guidance, infinite patience, encouragement and invaluable suggestions throughout my studies. I wish to express my immense gratitude to Professor Derek Bewley, Department of Botany, University of Guelph, Canada for his constructive criticism during the preparation of this thesis. I am also grateful to Drs. Frank Gubler and John V. Jacobsen, CSIRO Division of Plant Industry, Canberra for providing a cDNA library. I wish to thank Dr. Simon Robinson, Senior Scientist, CSIRO for his constructive suggestions throughout the duration of my research project. -

Stress-Triggered Activation of the Metalloprotease Oma1 Involves Its
University of Nebraska - Lincoln DigitalCommons@University of Nebraska - Lincoln Biochemistry -- Faculty Publications Biochemistry, Department of 2014 Stress-triggered Activation of the Metalloprotease Oma1 Involves Its C-terminal Region and Is Important for Mitochondrial Stress Protection in Yeast Iryna Bohovych University of Nebraska- Lincoln, [email protected] Garrett onD aldson University of Nebraska- Lincoln Sara Christianson University of Nebraska-Lincoln Nataliya Zahayko University of Nebraska-Lincoln Oleh Khalimonchuk University of Nebraska-Lincoln, [email protected] Follow this and additional works at: http://digitalcommons.unl.edu/biochemfacpub Part of the Biochemistry Commons, Biotechnology Commons, and the Other Biochemistry, Biophysics, and Structural Biology Commons Bohovych, Iryna; Donaldson, Garrett; Christianson, Sara; Zahayko, Nataliya; and Khalimonchuk, Oleh, "Stress-triggered Activation of the Metalloprotease Oma1 Involves Its C-terminal Region and Is Important for Mitochondrial Stress Protection in Yeast" (2014). Biochemistry -- Faculty Publications. 167. http://digitalcommons.unl.edu/biochemfacpub/167 This Article is brought to you for free and open access by the Biochemistry, Department of at DigitalCommons@University of Nebraska - Lincoln. It has been accepted for inclusion in Biochemistry -- Faculty Publications by an authorized administrator of DigitalCommons@University of Nebraska - Lincoln. THE JOURNAL OF BIOLOGICAL CHEMISTRY VOL. 289, NO. 19, pp. 13259–13272, May 9, 2014 © 2014 by The American Society -

Genomic and Proteomic Analyses of the Coral Pathogen Vibrio Coralliilyticus Reveal a Diverse Virulence Repertoire
The ISME Journal (2011) 5, 1471–1483 & 2011 International Society for Microbial Ecology All rights reserved 1751-7362/11 www.nature.com/ismej ORIGINAL ARTICLE Genomic and proteomic analyses of the coral pathogen Vibrio coralliilyticus reveal a diverse virulence repertoire Eidy de O Santos1, Nelson Alves Jr1, Graciela M Dias1, Ana Maria Mazotto2, Alane Vermelho2, Gary J Vora3, Bryan Wilson4, Victor H Beltran4, David G Bourne4, Fre´de´rique Le Roux5,6 and Fabiano L Thompson1 1Laboratory of Microbiology, Institute of Biology, Federal University of Rio de Janeiro (UFRJ), Rio de Janeiro, Brazil; 2Laboratory of Proteases, Institute of Microbiology, UFRJ, Rio de Janeiro, Brazil; 3Center for Bio/ Molecular Science & Engineering, Naval Research Laboratory, Washington, DC, USA; 4Centre for Marine Microbiology and Genetics, Australian Institute of Marine Science, Townsville MC, Queensland, Australia; 5FR2424 CNRS UPMC Station Biologique de Roscoff, France and 6Ifremer, Laboratoire de Physiologie des Inverte´bre´s, Brest, France Vibrio coralliilyticus has been implicated as an important pathogen of coral species worldwide. In this study, the nearly complete genome of Vibrio coralliilyticus strain P1 (LMG23696) was sequenced and proteases implicated in virulence of the strain were specifically investigated. The genome sequence of P1 (5 513 256 bp in size) consisted of 5222 coding sequences and 58 RNA genes (53 tRNAs and at least 5 rRNAs). Seventeen metalloprotease and effector (vgrG, hlyA and hcp) genes were identified in the genome and expressed proteases were also detected in the secretome of P1. As the VcpA zinc-metalloprotease has been considered an important virulence factor of V. coralliilyticus, a vcpA deletion mutant was constructed to evaluate the effect of this gene in animal pathogenesis. -

Role of PITRM1 in Mitochondrial Dysfunction and Neurodegeneration
biomedicines Review Role of PITRM1 in Mitochondrial Dysfunction and Neurodegeneration Dario Brunetti 1,2 , Alessia Catania 2, Carlo Viscomi 3, Michela Deleidi 4, Laurence A. Bindoff 5,6, Daniele Ghezzi 2,7,* and Massimo Zeviani 8,9,* 1 Department of Medical Biotechnology and Translational Medicine, University of Milan, 20129 Milan, Italy; [email protected] 2 Unit of Medical Genetics and Neurogenetics, Fondazione IRCCS Istituto Neurologico Carlo Besta, 20126 Milan, Italy; [email protected] 3 Department of Biomedical Sciences, University of Padova, 35131 Padova, Italy; [email protected] 4 German Center for Neurodegenerative Diseases (DZNE), 72076 Tübingen, Germany; [email protected] 5 Neuro-SysMed, Center of Excellence for Clinical Research in Neurological Diseases, Haukeland University Hospital, N-5021 Bergen, Norway; [email protected] 6 Department of Clinical Medicine, University of Bergen, N-5021 Bergen, Norway 7 Department of Pathophysiology and Transplantation, University of Milan, 20122 Milan, Italy 8 Department of Neurosciences, University of Padova, 35128 Padova, Italy 9 Venetian Institute of Molecular Medicine, 35128 Padova, Italy * Correspondence: [email protected] (D.G.); [email protected] (M.Z.) Abstract: Mounting evidence shows a link between mitochondrial dysfunction and neurodegenera- tive disorders, including Alzheimer Disease. Increased oxidative stress, defective mitodynamics, and impaired oxidative phosphorylation leading to decreased ATP production, can determine synaptic dysfunction, apoptosis, and neurodegeneration. Furthermore, mitochondrial proteostasis and the Citation: Brunetti, D.; Catania, A.; protease-mediated quality control system, carrying out degradation of potentially toxic peptides Viscomi, C.; Deleidi, M.; Bindoff, L.A.; and misfolded or damaged proteins inside mitochondria, are emerging as potential pathogenetic Ghezzi, D.; Zeviani, M. -

Composition Containing Protease Produced by Vibrio and Method of Use in Debridement and Wound Healing
Europaisches Patentamt European Patent Office 0 Publication number: 0 472 011 A1 Office europeen des brevets EUROPEAN PATENT APPLICATION 0 Application number: 91112732.2 int. CI 5: A61K 37/48, C12N 9/52, C12N 15/31, //(C12N 15/31, 0 Date of filing: 29.07.91 C12R1:63) 0 Priority: 15.08.90 US 567884 0 Inventor: Fortney, Donald Zane 13.03.91 US 670612 2114 Reese Road Westminster, Md. 21157(US) 0 Date of publication of application: Inventor: Durham, Donald Richard 26.02.92 Bulletin 92/09 20617 Beaver Ridge Road Gaithersburg, Md. 20879(US) 0 Designated Contracting States: AT BE CH DE DK ES FR GB GR IT LI LU NL SE 0 Representative: UEXKULL & STOLBERG 0 Applicant: W.R. Grace & Co.-Conn. Patentanwalte Grace Plaza, 1114 Ave. of the Americas Beselerstrasse 4 New York New York 10036(US) W-2000 Hamburg 52(DE) 0 Composition containing protease produced by vibrio and method of use in debridement and wound healing. 0 A composition for treating wounds comprising at least one pharmaceutically acceptable carrier admixed with an effective amount of a protease selected from the group consisting of: (a) an extracellular neutral protease produced by cultivation of a microorganism belonging to the genus Vibrio, said protease characterized by the following properties: i. hydrolyzes components of necrotic tissue including denatured collagen, elastin and fribrin; ii. does not substantially hydrolyze native tissue in viva, and iii. exhibits stable activity when stored at 25° C in a topical formulation; (b) a protease expressed by recombinant host cells which have been transformed or transfected with an expression vector for said protease (a); and (c) mutants and hybrids of proteases (a) and (b) which are characterized by the properties (i) to (iii).