This Is Normal Text
Total Page:16
File Type:pdf, Size:1020Kb

Load more
Recommended publications
-

For a Hero—The Silver Star Jury Tomorrow in the Cemetery Police Laboratory, in Trenton
Weather Dfctrilmtfc* 7 •jn, te«p#r*tu/» ». OrMr May wtt* Wchin the Mi. Clttr THEDAILY *and ood tonight with few in «s. 24,800 Tomorrow suitny and cool with 7 /ted Ban*; Area J high to the 50s. Wednesday fair •ad continued cool. NORTHERN MONMOUTH'S HOME NEWSPAPER DIAL 7414)010 dtli#. M«»|U» throo«h #rtd>r. l*ooa<l Cim Poiugt VOL. 87, NO. 81 Paid at AJdlifi andat AdijUooal Uallln( OUIcei. MONDAY, OCTOBER 19, 1964 7c PER COPY PAGE ONE Cemetery Case Goes Middletown Woman's Husband Killed in Viet Nam Before Grand Jury HOLMDEL — Mrs. Conrad A group of citizens found five Hess, South Laurel Ave., and human bones *- more than 100 Mrs. Marion Norton, Main St., years old in a mound of dirt both informed The Register that next to the excavation. The iden- they will appear before the Grand tification was made by the state For a Hero—The Silver Star Jury tomorrow in the cemetery police laboratory, in Trenton. case. Both Mrs. Hess and Mrs. Nor- WEST POINT, N. Y.—Mrs. Harriet L. Hines,- encouraged them to pursue their efforts in the At the decorations' ceremony here, another Both said they had received ton claim that they have fore- 186 Cherry Tree La., Middletown, N. J., has re- defense of their homeland." fallen hero, the late Capt. James P. Spruill, Suffern, mbpoenas to appear before the bears buried In the old ceme- jury. tery. ceived, with pride, though in grief, the Silver Star A Daily Register editorial April 28, noted N. Y., was honored with the Legion of Merit. -

Tobacco Labelling -.:: GEOCITIES.Ws
Council Directive 89/622/EC concerning the labelling of tobacco products, as amended TAR AND NICOTINE CONTENTS OF THE CIGARETTES SOLD ON THE EUROPEAN MARKET AUSTRIA Brand Tar Yield Nicotine Yield Mg. Mg. List 1 A3 14.0 0.8 A3 Filter 11.0 0.6 Belvedere 11.0 0.8 Camel Filters 14.0 1.1 Camel Filters 100 13.0 1.1 Camel Lights 8.0 0.7 Casablanca 6.0 0.6 Casablanca Ultra 2.0 0.2 Corso 4.0 0.4 Da Capo 9.0 0.4 Dames 9.0 0.6 Dames Filter Box 9.0 0.6 Ernte 23 13.0 0.8 Falk 5.0 0.4 Flirt 14.0 0.9 Flirt Filter 11.0 0.6 Golden Smart 12.0 0.8 HB 13.0 0.9 HB 100 14.0 1.0 Hobby 11.0 0.8 Hobby Box 11.0 0.8 Hobby Extra 11.0 0.8 Johnny Filter 11.0 0.9 Jonny 14.0 1.0 Kent 10.0 0.8 Kim 8.0 0.6 Kim Superlights 4.0 0.4 Lord Extra 8.0 0.6 Lucky Strike 13.0 1.0 Lucky Strike Lights 9.0 0.7 Marlboro 13.0 0.9 Marlboro 100 14.0 1.0 Marlboro Lights 7.0 0.6 Malboro Medium 9.0 0.7 Maverick 11.0 0.8 Memphis Classic 11.0 0.8 Memphis Blue 12.0 0.8 Memphis International 13.0 1.0 Memphis International 100 14.0 1.0 Memphis Lights 7.0 0.6 Memphis Lights 100 9.0 0.7 Memphis Medium 9.0 0.6 Memphis Menthol 7.0 0.5 Men 11.0 0.9 Men Light 5.0 0.5 Milde Sorte 8.0 0.5 Milde Sorte 1 1.0 0.1 Milde Sorte 100 9.0 0.5 Milde Sorte Super 6.0 0.3 Milde Sorte Ultra 4.0 0.4 Parisienne Mild 8.0 0.7 Parisienne Super 11.0 0.9 Peter Stuyvesant 12.0 0.8 Philip Morris Super Lights 4.0 0.4 Ronson 13.0 1.1 Smart Export 10.0 0.8 Treff 14.0 0.9 Trend 5.0 0.2 Trussardi Light 100 6.0 0.5 United E 12.0 0.9 Winston 13.0 0.9 York 9.0 0.7 List 2 Auslese de luxe 1.0 0.1 Benson & Hedges 12.0 1.0 Camel 15.0 1.0 -

May 2013 ANISHINABEK NEWS the Voice of the Anishinabek Nation
Page 1 Anishinabek News May 2013 ANISHINABEK NEWS The voice of the Anishinabek Nation Volume 25 Issue 4 Published monthly by the Union of Ontario Indians - Anishinabek Nation Single Copy: $2.00 May 2013 OUR TIME IS NOW: Madahbee UOI Offi ces –This year marks the resentative of the Crown back then, governments that we don’t need in April, 2014. 250th anniversary of The Royal there was no doubt that we were ca- them to tell us how to look after our Madahbee pointed to the recent Proclamation, a landmark document pable of managing our own affairs, citizens.” stream of Harper government legis- by which the British Crown recog- governing our own communities – A key agenda item at the June lation that ignores First Nations ju- nized the nationhood of “the Indian the British representatives were well 3-6 assembly in Munsee-Delaware risdiction on everything from First tribes of North America.” aware of our ability to assert our ju- will be the future of the Anishinabek Nations election processes to matri- Commemoration of the 1763 risdiction because, when push came Education System, which is intend- monial real property rights. event is an agenda item for June’s to shove, First Nation warriors dev- ed to restore jurisdiction for educa- “We need to start occupying the annual general assembly of An- astated nine of eleven British Forts tion of some 2500 K-12 Anishina- fi eld in these areas – we’ve got to get doyouknowyourrights.ca ishinabek Nation chiefs, and Grand in Pontiac’s War. bek Nation students. Half a day on moving on some of these issues. -

23/F, 8-Commercial Tower, 8 Sun Yip Street, Chai Wan, Hong Kong Tel 25799398 26930136 Fax (+852) 26027153 Email [email protected]
To Whom it may concern ISO Test methods for cigarette tar and nicotine content are outdated and unrepresentative of the actual yield and toxins intake due to smoker compensation - Countries should adopt the Health Canada Intense test method, like RIVM Holland The old and outdated ISO test criteria for cigarette tar and nicotine content used by the HK Government Lab is way out of date. The industry deliberately perforates the filter and paper of the tobacco rods with tiny holes to ‘cheat’ the current ISO machine test methods. What actually happens is the smokers wrap their fingers and of course mouth around the filter to compensate for the additional dilution air being sucked in through the perforations. The ISO smoking test machine is not real world, does not compensate by blocking the holes and hence reveals test results that are far, far lower than the smokers actually inhale. RIVM, the Dutch Ministry of Health, has adopted the Health Canada Intense smoking test criteria which better reveals the actual tar and nicotine in each cigarette rod since they tape over the perforated holes in the same way that the smoker compensates with fingers and mouth, to seal the holes - and then test the actual values. Attached herewith you can see the vast disparities as revealed in the RIVM test data which show the level of toxics which the smokers actually inhale versus the mythical ISO data preferred and provided by the manufacturers. Countries Kong need to switch to the Health Canada Intense method of cigarette testing asap and inform the public accordingly of the actual level of toxins they inhale when they smoke cigarettes. -

First Name Last Name Member # Anne Aamot 75275 Callie Aamot
This is the list of Qualifiers for the 2021 Open World Show. If your birthday is January 2, 2001 or later you are also eligible to attend the 2021 Youth World Show. First Name Last Name Member # Anne Aamot 75275 Callie Aamot 87020 Candice Aamot 96851 Brittany Aaron 46770 Raynell Aaron 4984 Stephanie Aas 81069 Blake Abadie 101834 Danielle Abbate 108459 Bryndalynn Abbott 102102 Jennifer Abbott 106774 Sarah Abbott 73410 Fawna Lee Abel 109166 Jessica Abel 24224 Kylie Abell 108788 Mackenzie Abercrombie 84925 Charlene Aberg 89812 Mary Ella Abernathy 106105 Tiffany Abernathy 105675 Chloe Abfalter 107816 Keily Abney 77484 Abbey Abraham 108015 Dallin Abraham 107460 Kelli Abraham 107459 Krista Abrahamson 101115 Renn Abramczyk - Dubiel 83363 Jadyn Abrams 105281 Pamela Abrams 1700 Kaitlyn Absatz 90283 Crystal Ackerman 79023 Natalia Ackerman 103140 Ella Ackley 92283 Anna Acopian 6728 Tallulah Rae Acopian 106651 Adrienne Acord 106723 Josie Acord 84515 Gloria Acosta 96682 Kimberly Acosta 31653 Tim Acosta 74327 Page 1 This is the list of Qualifiers for the 2021 Open World Show. If your birthday is January 2, 2001 or later you are also eligible to attend the 2021 Youth World Show. First Name Last Name Member # Rebecca Acre 106888 Isabella Actis 106278 Debbie Acuff 8595 Courtney Adair 49650 Paulette Adair 14971 Todd Adair 28191 Shayla Adam 91381 Alaina Adams 103253 Alexandra Adams 60114 Amanda Adams 86187 Amber Adams 72570 Angela Adams 75503 Angelina Adams 103254 Aspen Adams 105506 Ava Adams 109404 Beth Adams 18895 Candice Adams 79385 Cassie Adams 107259 Dee Adams 8117 Dorothy Adams 4794 Graan Adams 106853 Grace Adams 90768 Gracen K Adams 108477 Haley Adams 70207 Haley Adams 107087 Hannah Adams 86188 Jenny Adams 2291 Jessica Adams 96790 Julia Adams 45142 Karleigh Adams 89574 Kaydee Adams 102435 Kaylie D Adams 108478 Kelly Adams 36681 Khloe Adams 105882 Lilly Adams 81628 Lynlee Adams 108969 Morgan Adams 65619 Remi Adams 108092 Page 2 This is the list of Qualifiers for the 2021 Open World Show. -

NVWA Nr. Merk/Type Nicotine (Mg/Sig) NFDPM (Teer)
NVWA nr. Merk/type Nicotine (mg/sig) NFDPM (teer) (mg/sig) CO (mg/sig) 89537347 Lexington 1,06 12,1 7,6 89398436 Camel Original 0,92 11,7 8,9 89398339 Lucky Strike Original Red 0,88 11,4 11,6 89398223 JPS Red 0,87 11,1 10,9 89399459 Bastos Filter 1,04 10,9 10,1 89399483 Belinda Super Kings 0,86 10,8 11,8 89398347 Mantano 0,83 10,8 7,3 89398274 Gauloises Brunes 0,74 10,7 10,1 89398967 Winston Classic 0,94 10,6 11,4 89399394 Titaan Red 0,75 10,5 11,1 89399572 Gauloises 0,88 10,5 10,6 89399408 Elixyr Groen 0,85 10,4 10,9 89398266 Gauloises Blondes Blue 0,77 10,4 9,8 89398886 Lucky Strike Red Additive Free 0,85 10,3 10,4 89537266 Dunhill International 0,94 10,3 9,9 89398932 Superkings Original Black 0,92 10,1 10,1 89537231 Mark 1 New Red 0,78 10 11,2 89399467 Benson & Hedges Gold 0,9 10 10,9 89398118 Peter Stuyvesant Red 0,82 10 10,9 89398428 L & M Red Label 0,78 10 10,5 89398959 Lambert & Butler Original Silver 0,91 10 10,3 89399645 Gladstone Classic 0,77 10 10,3 89398851 Lucky Strike Ice Gold 0,75 9,9 11,5 89537223 Mark 1 Green 0,68 9,8 11,2 89393671 Pall Mall Red 0,84 9,8 10,8 89399653 Chesterfield Red 0,75 9,8 10 89398371 Lucky strike Gold 0,76 9,7 11,1 89399599 Marlboro Gold 0,78 9,6 10,3 89398193 Davidoff Classic 0,88 9,6 10,1 89399637 Marlboro Red 100 0,79 9,6 9 89398878 Lucky Strike Ice 0,71 9,5 10,9 89398045 Pall Mall Red 0,81 9,5 10,5 89398355 Dunhill Master Blend Red 0,82 9,5 9,8 89399475 JPS Red 0,81 9,5 9,6 89398029 Marlboro Red 0,79 9,5 9 89537355 Elixyr Red 0,79 9,4 9,8 89398037 Marlboro Menthol 0,72 9,3 10,3 89399505 Marlboro -

Tie WESTFIELD LEADER
1- o >- !-• - tiE WESTFIELD LEADER •-I t.i i-i WJtfrfj, Clrtutmted H>**7* Nwmmr It Umim* Cmmmi* Published _..t H YEAR, NO. 4J WESTFIELD, NEW JERSEY, THURSDAY, JUNE 6, IMS 24 Pages—30 Cents (-i Every Thursday Only 17% of Voters Board Approves Sick Turn Out at Primary Wattietd's 21 poUing booths at Repubttcan-centreUed hoard cur- John Russo, 11«; Newark tractod an average of only 11 rently chaired by Malgran. Mayor Kenneth Gibson, 1*»; Leave, Resignation voten per hour Tuesday in one of Eliciting possibly the greatest Elliot Greenspan, 30; Stephen the lightest Primary Election The Westfield Board of Educa- 14 by an undercover state police Mrs. Moran stated that she was interest in Tuesday's election Wiley, 10»; and Robert Del Tufo, tion approved a sick leave exten- detective at the Madison Hill "against the procedure in this contests on record. Only 3,W7 was the race for the Democratic 47. residents, of more than It.oea nod te face the incumbent ding to Dec. 31,1M», for Samuel Rest Area on the Garden State case." Mr. Weimer concurred, registered to vote, cast ballots. A. Soprano and accepted his Parkway. A decision on the case saying they were "rewarding Republican governor in Both local political parties will resignation effective that date at will be made in about two weeks, something that shouldn't be re- While no contests were en the November. As voters did organize next Monday with eom- a special meeting Tuesday. according to Municipal warded." Republican ballot, l,«l from statewide, local residents gave mitteemen and women elected that political party went to the Peter Shapiro a wide margin of Dr. -
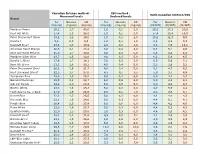
TNCO Levels and Ratio's
Canadian Intense method - ISO method - Ratio Canadian Intense/ISO Measured levels Declared levels Brand Tar Nicotine CO Tar Nicotine CO Tar Nicotine CO (mg/cig) (mg/cig) (mg/cig) (mg/cig) (mg/cig) (mg/cig) (CI/ISO) (CI/ISO) (CI/ISO) Marlboro Prime 26,1 1,7 40,0 1,0 0,1 2,0 26,1 17,2 20,0 Kent HD White 17,4 1,3 28,0 1,0 0,1 2,0 17,4 13,4 14,0 Peter Stuyvesant Silver 15,2 1,2 19,0 1,0 0,1 2,0 15,2 12,3 9,5 Karelia I 9,6 0,9 9,3 1,0 0,1 1,0 9,6 8,6 9,3 Davidoff Blue* 23,6 1,7 30,9 2,9 0,2 2,6 8,3 7,0 12,1 American Spirit Orange 20,5 2,1 18,4 3,0 0,4 4,0 6,8 5,1 4,6 Kent Surround Menthol 25,0 1,7 30,8 4,0 0,4 5,0 6,3 4,3 6,2 Marlboro Silver Blue 24,7 1,5 32,6 4,0 0,3 5,0 6,2 5,0 6,5 Karelia L (Blue) 17,6 1,7 14,1 3,0 0,3 2,0 5,9 5,6 7,1 Kent HD Silver 21,1 1,6 26,2 4,0 0,4 5,0 5,3 3,9 5,2 Peter Stuyvesant Blue* 20,2 1,6 21,7 4,0 0,4 5,0 5,1 4,7 4,3 Kent Surround Silver* 22,5 1,7 27,0 4,5 0,5 5,5 5,0 3,7 4,9 Templeton Blue 25,0 1,8 26,2 5,0 0,4 6,0 5,0 4,4 4,4 Belinda Filterkings 29,9 2,2 24,7 6,0 0,5 6,0 5,0 4,3 4,1 Silk Cut Purple 24,9 2,0 23,4 5,0 0,5 5,0 5,0 4,0 4,7 Boston White 23,3 1,6 25,3 5,0 0,3 6,0 4,7 5,5 4,2 Mark Adams No. -
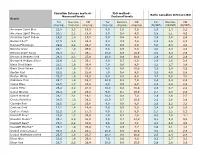
TNCO Levels and Ratio's
Canadian Intense method - ISO method - Ratio Canadian Intense/ISO Measured levels Declared levels Brand Tar Nicotine CO Tar Nicotine CO Tar Nicotine CO (mg/cig) (mg/cig) (mg/cig) (mg/cig) (mg/cig) (mg/cig) (CI/ISO) (CI/ISO) (CI/ISO) American Spirit Blue 22,8 2,3 19,5 9,0 1,0 9,0 2,5 2,3 2,2 American Spirit Orange 20,5 2,1 18,4 3,0 0,4 4,0 6,8 5,1 4,6 American Spirit Yellow 19,5 1,8 17,0 5,0 0,6 6,0 3,9 3,0 2,8 Bastos Filter* 27,9 2,3 22,3 9,8 0,9 7,6 2,8 2,5 2,9 Belinda Filterkings 29,9 2,2 24,7 6,0 0,5 6,0 5,0 4,3 4,1 Belinda Green 24,1 1,9 25,6 6,0 0,5 6,0 4,0 3,9 4,3 Belinda Super Kings 36,3 2,7 29,5 10,0 0,8 10,0 3,6 3,4 2,9 Benson & Hedges Gold 28,3 2,3 27,9 10,0 0,9 10,0 2,8 2,6 2,8 Benson & Hedges Silver 22,6 1,8 25,1 8,0 0,7 9,0 2,8 2,5 2,8 Black Devil Black 23,1 1,6 30,4 7,0 0,6 9,0 3,3 2,7 3,4 Black Devil Yellow 25,4 1,8 31,6 8,0 0,6 10,0 3,2 2,9 3,2 Boston Red 23,2 1,6 25,4 7,0 0,4 9,0 3,3 4,0 2,8 Boston White 23,3 1,6 25,3 5,0 0,3 6,0 4,7 5,5 4,2 Caballero Plain 23,7 1,9 15,8 10,0 0,8 7,0 2,4 2,3 2,3 Camel Blue 22,5 1,7 23,8 8,0 0,6 9,0 2,8 2,8 2,6 Camel Filter 26,2 2,2 21,9 10,0 0,8 10,0 2,6 2,7 2,2 Camel Orange 24,4 1,9 23,6 9,0 0,7 10,0 2,7 2,8 2,4 Camel Original 28,1 2,1 19,1 10,0 0,8 7,0 2,8 2,6 2,7 Chesterfield Red 26,5 1,8 28,2 10,0 0,7 10,0 2,6 2,6 2,8 Claridge Red 22,6 1,3 26,6 8,0 0,6 8,0 2,8 2,2 3,3 Couture Gold 17,1 1,4 14,4 5,0 0,5 5,0 3,4 2,9 2,9 Couture Purple 21,2 1,8 17,3 8,0 0,7 8,0 2,7 2,5 2,2 Davidoff Blue* 23,6 1,7 30,9 2,9 0,2 2,6 8,3 7,0 12,1 Davidoff Classic* 29,1 2,4 31,0 9,5 0,7 10,4 3,1 3,3 3,0 Davidoff -

The Hillside Times, Friday, July 6, 1984 (USPS 245-780) 923 9207 Strike Underlines Board and Twsp
WEATHER (Provided by the National Weather Service) FRIDA Y-Occasional showers and thundershowers. Heavy downpours at times. Warm and humid, in 80 s. t& \\t ^Ulatfo ®tm?H SATURDAY-Chance of early shower. Partly sunny, less humid, in 80 ’ s. SUNDAY-MONDAY-Falr, Serving Hillside Since 1924 less humid. High 80-85. VOL 58 NO 29 The Hillside Times, Friday, July 6, 1984 (USPS 245-780) 923 9207 Strike Underlines Board And Twsp. Committee At Odds Waste Disposal Problems Over Welfare Director Appointment In a repeat of a similar The town's plan would per At Tuesday's Committee By W alt Berthold May meeting, by a 3-0 vote, members with no legal as strike In 1981, Garbage dis mit removal of recyclable ma session a resolution noting the claimed she was entitled to portedly occupying the W el with two members, Including sistance available from the posal workers throughout terials, but residents would health hazards and a possible handle the office until a new fare Department office, and The controversy over the Township Committeeman Township to handle their side most of northern and central still have to store organic explosion" In Insect and ro permanent appointment could the Board refuses to name naming of a new Welfare Di Anthony Deo and Mrs. Davis, of the dispute. New Jesey went out on strike waste, hopefully with dent Infestation, forbid the be made. another Acting Director pend rector for Hillside continued abstaining. The Committee, by a 4-1 this week, closing down dis measures that will reduce placement of waste at curb- This memorandum was ing the hearing. -

Badgercare Reform Demonstration Waiver Extension – Draft Application
State of Wisconsin BadgerCare Reform Demonstration Project Draft 1115 Waiver Extension Application December 5, 2017 State of Wisconsin, Department of Health Services BadgerCare Reform Demonstration Project Draft 1115 Waiver Extension Application Table of Contents 1.0 Introduction ............................................................................................................................... 1 2.0 Historical Narrative and Program Description ......................................................................... 2 3.0 Program Changes ...................................................................................................................... 6 4.0 Waiver and Expenditure Authorities ........................................................................................ 7 5.0 Quality Monitoring ................................................................................................................... 9 6.0 Budget Neutrality and Monitoring .......................................................................................... 10 7.0 Demonstration Evaluation ...................................................................................................... 12 8.0 Public Notice Process ............................................................................................................. 13 Appendix A – Abbreviated Public Notice Appendix B – Long Public Notice Appendix C – Section 1115 Demonstration Waiver Amendment Application Appendix D – Section 1115 Demonstration Waiver for Medicaid Coverage -

Board of Selectmen Move to Enforce Oil Ordinance
n,.O..i,:..?} imiOivIi.L- LIBiua^Y T. rT Il..VKi*, GT. "t Thursday, March 23, 1950 THE BRANFORD REVIEW - EAST HAVEN NEWS Page Eight to servo all departments In handl-, Ing common functions of personnel, WHAT EAST HAVEN BOOSTS I i public works and gonsral admlnl."!- G.O.P. Women North Branford CLASSIFIED ADS DEMOCRATIC WOMEN trativo services. BOOSTS EAST HAVEN! 4. Fixing clear responsibility for CONGREOA-nONAL CnURCH bettor management on the Chief Hear Writer Kcv. B. C. Trent, Pastor HELP WANTED SITUATIONS WANTED MAKE EAST HAVEN A BIGGER, HEAR LENIHAN TALK Executive 1 and providing him with Miss Ethel Maynard the meons of discharging It. Organist and Choir Director BUY - RENT SELL - HAVE IT REPAIRED BETTER, BUSIER COMMUNITY 5. Defining afresh the status and Talk Politics 11:00 Morning worship ON REORGANIZATION role of roads and commissions. 9:45 Sunday School WORDS FOUR Combined With The Branford Review 6. Providing more full and Inprcsslons of the Washington $1.50 M. J. tcnihan wns the guest Mr. Lcnlhnn outlined the pro- systematically for effective citizen scene as viewed by Mrs. Helen ZION EPISCOPAL CHURCH 25 or LESS 50f( TIMES B Conh Per Copy—Two DolUi't A Year Democratic Club meeting held last po.sals as follows: participation In the work of the Lombard, prominent newspaper Rev. Francis 1, Smith, Rector One T'imo EAST HAVEN, CONNECTICUT, THURSDAY, MARCH 30, 1950 fi flpcaker at the Federated Women's 1. Grouping all slate functions so departments and In the formula columnist of the nation's capltol, Edmund L, Stoddard VOL.