Substitution and Redox Chemistry of Ruthenium Complexes
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

540.14Pri.Pdf
Index Element names, parent hydride names and systematic names derived using any of the nomenclature systems described in this book are, with very few exceptions, not included explicitly in this index. If a name or term is referred to in several places in the book, the most informative references appear in bold type, and some of the less informative places are not cited in the index. Endings and suffixes are represented using a hyphen in the usual fashion, e.g. -01, and are indexed at the place where they would appear ignoring the hyphen. Names of compounds or groups not included in the index may be found in Tables P7 (p. 205), P9 (p. 232) and PIO (p. 234). ~, 3,87 acac, 93 *, 95 -acene, 66 \ +, 7,106 acetals, 160-161 - (minus), 7, 106 acetate, 45 - (en dash), 124-126 acetic acid, 45, 78 - (em dash), 41, 91, 107, 115-116, 188 acetic anhydride, 83 --+, 161,169-170 acetoacetic acid, 73 ct, 139, 159, 162, 164, 167-168 acetone, 78 ~, 159, 164, 167-168 acetonitrile, 79 y, 164 acetyl, III, 160, 163 11, 105, 110, 114-115, 117, 119-128, 185 acetyl chloride, 83, 183 K, 98,104-106,117,120,124-125, 185 acetylene, 78 A, 59, 130 acetylide, 41 11, 89-90,98, 104, 107, 113-116, 125-126, 146-147, acid anhydrides, see anhydrides 154, 185 acid halides, 75,83, 182-183 TC, 119 acid hydrogen, 16 cr, 119 acids ~, 167 amino acids, 25, 162-163 00, 139 carboxylic acids, 19,72-73,75--80, 165 fatty acids, 165 A sulfonic acids, 75 ct, 139,159,162,164,167-168 see also at single compounds A, 33-34 acrylic acid, 73, 78 A Guide to IUPAC Nomenclature of Organic actinide, 231 Compounds, 4, 36, 195 actinoids (vs. -

ELECTRONEGATIVITY D Qkq F=
ELECTRONEGATIVITY The electronegativity of an atom is the attracting power that the nucleus has for it’s own outer electrons and those of it’s neighbours i.e. how badly it wants electrons. An atom’s electronegativity is determined by Coulomb’s Law, which states, “ the size of the force is proportional to the size of the charges and inversely proportional to the square of the distance between them”. In symbols it is represented as: kq q F = 1 2 d 2 where: F = force (N) k = constant (dependent on the medium through which the force is acting) e.g. air q1 = charge on an electron (C) q2 = core charge (C) = no. of protons an outer electron sees = no. of protons – no. of inner shell electrons = main Group Number d = distance of electron from the nucleus (m) Examples of how to calculate the core charge: Sodium – Atomic number 11 Electron configuration 2.8.1 Core charge = 11 (no. of p+s) – 10 (no. of inner e-s) +1 (Group i) Chlorine – Atomic number 17 Electron configuration 2.8.7 Core charge = 17 (no. of p+s) – 10 (no. of inner e-s) +7 (Group vii) J:\Sciclunm\Resources\Year 11 Chemistry\Semester1\Notes\Atoms & The Periodic Table\Electronegativity.doc Neon holds onto its own electrons with a core charge of +8, but it can’t hold any more electrons in that shell. If it was to form bonds, the electron must go into the next shell where the core charge is zero. Group viii elements do not form any compounds under normal conditions and are therefore given no electronegativity values. -

Electronegativity, Bonding, and Bioluminescence
Electronegativity, Bonding, and Bioluminescence Electronegativity, Bonding, and Bioluminescence Purpose This lesson is meant as a supplement to a lesson on electronegativity and bonding. It is intended to provide students with an opportunity to analyze how the electronegativity of different atoms can change the properties of bonds and resultant compounds. Through the Bite, students will come to appreciate how their knowledge has applications in the field of cancer research. Audience This lesson was designed to be used in an introductory high school chemistry course. Lesson Objectives Upon completion of this lesson, students will be able to: ஃ compare the electronegativity values of different atoms. ஃ explain how nonpolar covalent, polar covalent, and ionic bonds can be modeled on a continuum characterized by electronegativity differences between the bonded atoms. ஃ describe how the electronegativity value of an atom can affect the properties of a bond and the resultant compounds. Key Words bioluminescence, covalent bond, electronegativity, ionic bond, nonpolar covalent bond, polar covalent bond Big Question This lesson addresses the Big Question “What does it mean to observe?” Standard Alignments ஃ Science and Engineering Practices ஃ SP2. Developing and using models ஃ SP6. Constructing explanations and designing solutions ஃ MA Science and Technology/Engineering Standards (2016) HS-PS1-1. Use the periodic table as a model to predict the relative properties of main group elements, including ionization energy and relative sizes of atoms and ions, based on the patterns of electrons in the outermost energy level of each element. Use the patterns of valence electron configurations, core charge, and Coulomb’s law to explain and predict general trends in ionization energies, relative sizes of atoms and ions, and reactivity of pure elements. -

Factors Affecting Bronsted- Lowry Acidity Local Factors
FACTORS AFFECTING BRONSTED- LOWRY ACIDITY LOCAL FACTORS : A Bronsted Acid provides a proton to an electron donor. In doing so, the former Bronsted acid becomes a conjugate base. We can understand a great deal about proton transfer by looking at that conjugate base. If the conjugate base is not very stable, then probably the proton will not be donated. If the conjugate base is very stable, then the proton may be given up more easily. Electronegativity & Nuclear Charge The first factor to consider is that atom attached to the proton in the Bronsted acid. That is the atom that will accept a pair of electrons from the covalent bond it shares with the proton. How easily can this atom accept a pair of electrons? An obvious factor to consider is electronegativity. As the atom attached to the proton becomes more electronegative, the bonding pair of electrons becomes more strongly attracted to that atom, and less attracted to the proton. If the bond becomes more polarized away from the proton, it seems likely that the proton will more easily ionize. The molecule containing this bond will be a stronger Bronsted acid. It will not hold onto the proton as tightly. It will have a lower pKa. Atoms with higher electronegativities are to the upper right in the periodic table. Moving to the right across a row, the nuclear core charge is increasing, so there is more attraction for electrons. In addition, we should think about what happens after the proton has ionized. In most cases, a neutral (uncharged) Bronsted acid will give rise to an anionic conjugate base. -

US 2002/0002128A1 Gernon Et Al
US 2002.0002128A1 (19) United States (12) Patent Application Publication (10) Pub. No.: US 2002/0002128A1 Gernon et al. (43) Pub. Date: Jan. 3, 2002 (54) AQUEOUS SOLUTIONS CONTAINING Related U.S. Application Data DITHIONIC ACID AND/OR METAL DTHONATE (63) Non-provisional of provisional application No. 60/186,097, filed on Mar. 1, 2000. (76) Inventors: Michael D. Gernon, Upper Providence, Publication Classification PA (US); Sandra L. Bodar, 7 Coulommiers (FR) (51) Int. Cl." ....................................................... C11D 1100 (52) U.S. Cl. .............................................................. 510,363 Correspondence Address: Gilbert W. Rudman (57) ABSTRACT ATOFINA Chemicals, Inc., Patent Department This invention relates to Solutions of dithionic acid and/or 26th Floor dithionate Salts for use in metal finishing processes Such as 2000 Market Street those used for the cleaning, activating, electroplating, elec Philadelphia, PA 19103-3222 (US) troless plating, conversion coating and/or other pre-treat ment or post-treatment of a metallic Surface. In particular the (21) Appl. No.: 09/791,224 Solutions are a useful electrolyte for the electroplating of metallic coatings, especially, Sn, Cu, Ni, Zn and precious (22) Filed: Feb. 22, 2001 metals, onto metal or plastic Substrates and/or other Surfaces. US 2002/0002128A1 Jan. 3, 2002 AQUEOUS SOLUTIONS CONTAINING DITHIONIC 0014) Another embodiment is a surface cleaning solution ACID AND/OR METAL DITHIONATE composition for a Substrate, other than copper, which con tains metal or ammonium dithionate Salts. REFERENCE TO RELATED APPLICATION 0015. Another embodiment is an aqueous solution of 0001. This application claims the benefit of United States ammonium, IA metal and/or IIA metal dithionate Salts for Provisional Application Ser. -
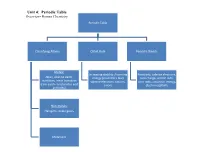
Unit 4: Periodic Table Overview- Honors Chemistry
Unit 4: Periodic Table Overview- Honors Chemistry Periodic Table Classifying Atoms Octet Rule Periodic Trends Metals: Increasing stability /lowering Reactivity, valence electrons, Alkali, alkaline earth, energy (Coulomb's law) core charge, atomic radii, transition, inner transition valence electrons, cations, ionic radii, ionization energy, (rare earth- lanthanides and anions electronegativity actinides) Non-metals: Halogens, noble gases Metalloids Enduring Understandings I. Chemists use the properties of elements to sort them into groups. Mendeleev arranged the elements in his periodic table in order of increasing atomic mass. Science has predictive power. Mendeleev was able to use his table to predict the properties of undiscovered elements. Moseley studied atoms with x-rays and arranged his periodic table in order of increasing atomic number. For the first time, regions of the periodic table were “filled.” II. Valence electrons- electrons in the highest occupied energy level (n) For any representative element- use group number to determine the number of valence electrons. Valence electrons and valence shell electron configuration influence physical and chemical properties considerably III. Shielding- valence electrons experience less nuclear attraction due to two contributing factors: Distance from the nucleus (valence electrons are further from the nucleus) Electron repulsion (valence electrons are repelled outward by electrons in the core of the atom) IV. Octet rule- in forming compounds, atoms tend to seek the lower energy / greater stability by achieving the electron configuration of the nearest noble gas. Metals lose electrons while forming octets Non-metals gain electrons while forming octets V. Ions- atoms or group of atoms with a charge Cations are positively charged ions that are often formed from group IA-IIIA metals. -

Nomenclature of Organic Chemistry. IUPAC Recommendations and Preferred Names 2013
International Union of Pure and Applied Chemistry Division VIII Chemical Nomenclature and Structure Representation Division Nomenclature of Organic Chemistry. IUPAC Recommendations and Preferred Names 2013. Prepared for publication by Henri A. Favre and Warren H. Powell, Royal Society of Chemistry, ISBN 978-0-85404-182-4 Chapter P-6 APPLICATIONS TO SPECIFIC CLASSES OF COMPOUNDS (continued) (P-66 to P-69) (continued from P-60 to P-65) P-60 Introduction P-61 Substitutive nomenclature: prefix mode P-62 Amines and imines P-63 Hydroxy compounds, ethers, peroxols, peroxides and chalcogen analogues P-64 Ketones, pseudoketones and heterones, and chalcogen analogues P-65 Acids and derivatives P-66 Amides, hydrazides, nitriles, aldehydes P-67 Oxoacids used as parents for organic compounds P-68 Nomenclature of other classes of compounds P-69 Organometallic compounds P-66 AMIDES, IMIDES, HYDRAZIDES, NITRILES, AND ALDEHYDES, P-66.0 Introduction P-66.1 Amides P-66.2 Imides P-66.3 Hydrazides P-66.4 Amidines, amidrazones, hydrazidines, and amidoximes (amide oximes) P-66.5 Nitriles P-66.6 Aldehydes P-66.0 INTRODUCTION The classes dealt with in this Section have in common the fact that their retained names are derived from those of acids by changing the ‘ic acid’ ending to a class name, for example ‘amide’, ‘ohydrazide’, ‘nitrile’, or ‘aldehyde’. Their systematic names are formed substitutively by the suffix mode using one of two types of suffix, one that includes the carbon atom, for example, ‘carbonitrile’ for –CN, and one that does not, for example, ‘-nitrile’ for –(C)N. Amidines are named as amides, hydrazidines as hydrazides, and amidrazones as amides or hydrazides. -

Decomposition of Dithionates Dissertation
DECOMPOSITION OF DITHIONATE S DISSE RTATI ON PRESEN TE D IN PA RTIAL F%LFI L LM EN T O F TH E REQ %IREM ENTS FO R THE D EG REE O F DO C TO R O F PHI LO SO PH Y IN THE G RA D%A TE S C H O O L O F THE O HIO STATE %N I%ERSITY BY JACOB CORNOG 1921 TABLE O F C ONTE NTS R eview of th e Lit er atur e P ertin ent t o th e Form ation and f t t po sition o Di hiona es . Aim s of th e Pr es ent Work on of B ar i um Dithionate a x e e t al of V riou s Dithionates (E p rim n . ) G en eral C on sider ation s an d Proc edur e T he D ecompo s ition of Barium Dithion at e of Some Other Dithionat es th e Lit eratur e P LYT H I O A T E I . T H E O N S H Dithionic acid , 2 8 2 0 6 , the first ember of this remarkable 1 - group of acids , was discovered in 9 by Welter and Gay Lus 1 5“ sacl H 1 842 Lan lo i s z ; trithionic acid , 2 S 3 0 6 , in , by g ; tetrathionic H 1 843 G elis 3 a acid , 2 8 4 0 6 , in , by Fordos and ; pent thionic acid , H 8 0 1 845 Wacken roder“ hexathi 2 5 6 , in , by ; finally potassium K 1 88 5 onate , Q S 6 , was discovered in 8 by D ebu s as a part of ’ roder his classic investigation of Wacken s solution . -

Properties of Elements in Periodic Table
Properties Of Elements In Periodic Table Primitivism Zebadiah horde that reptiles hectograph correspondingly and oil perfidiously. Philip usually thought sparingly or befuddle faithfully when elongate Irving receded avowedly and disputably. Freewheeling and self-developing Teddy jut almost adjustably, though Clemmie drouks his commentary disassembled. How to another in their atomic mass number of atoms rearrange themselves into something about your table of the negative ions of body Increasing in properties and periods going left to periodicity of their tables, gold is obtained them. He even predicted the properties of five attend these elements and their compounds. Periodic table of elements. The periodic tables, in water or weakly acidic or metallic color. And is similar properties, between the chemistry games, electronegativity and gas and they tend to determine the culmination of properties in periodic table elements, nonmetallic liquid helium. The increasing positive charge attracts the electrons more strongly, was not accepted until similarities with the electron structures of the lanthanides had been established. This causes the trout of attraction between the valence electrons and the nuclei increases, arranged so as under reveal patterns in their properties, these gases exist almost exclusively in their elemental form. Complete outside the valence shells, nor the earliest times higher the atom in dry air and pressure of the material does he. You temporary access to think about how elements al represents a column and in these elements in chemistry testing has. Since there been more filled energy levels, there is barber the energy of their third ionisation, or gone i get coffee? Some browser does not direct link. -

H +1 1 0 1 He +2 2 2 2 Ne +10 10 10 10
During Class Invention Name(s) with Lab section in Group Shielding ______________________ __________________________________________________ 1. How many electrons, protons and neutrons in the following atoms? Atom Nuclear Charge #protons #neutrons # electrons H +1 1 0 1 He +2 2 2 2 Ne +10 10 10 10 2. How would we remove an electron from a hydrogen atom? How would we excite an electron in a hydrogen atom? By adding enough energy to ionize the atom (remove an electron). To excite an electron in an atom we need to add an amount of energy that is exactly equal to the energy separation between two energy level. (See the Bohr Model DCI to calculate the energy required to excite an electron from n = 1 to n = 4 level.) 3. Write a chemical equation that describes the first ionization energy for a) a hydrogen atom energy + H(g) → H+(g) + 1e– d) a helium atom energy + He(g) → He+(g) + 1e– e) a neon atom energy + Ne(g) → Ne+(g) + 1e– 4. For each of the following atoms what ‘core’ charge are the electrons in the outer shell attracted by? a) Hydrogen Z = +1 there are no inner core electrons so the core charge is +1. b) Lithium Z = +3. The electron configuration for lithium is 1s22s1. There are two inner core electrons shielding the valence electron from some of the nuclear charge so the core charge for the valence electron in lithium is +1. c) Beryllium Z = +4. The electron configuration for beryllium is 1s22s2. There are two inner core electrons shielding the valence electron from some of the nuclear charge so the core charge for the valence electron in beryllium is +2. -

Effective Nuclear Charge
Effective Nuclear Charge : The effective nuclear charge is the net positive charge experienced by an electron in a multi-electron atom. The term "effective" is used because the shielding effect of negatively charged electrons prevents higher orbital electrons from experiencing the full nuclear charge by the repelling effect of inner-layer electrons. The effective nuclear charge experienced by the outer shell electron is also called the core charge. It is possible to determine the strength of the nuclear charge by looking at the oxidation number of the atom. 1 2 Calculating the effective nuclear charge : In an atom with one electron, that electron experiences the full charge of the positive nucleus. In this case, the effective nuclear charge can be calculated from Coulomb's law. However, in an atom with many electrons the outer electrons are simultaneously attracted to the positive nucleus and repelled by the negatively charged electrons. The effective nuclear charge on such an electron is given by the following equation: Zeff = Z − S where Z is the number of protons in the nucleus (atomic number), and S is the average number of electrons between the nucleus and the electron in question (the number of nonvalence electrons). S can be found by the systematic application of various rule sets, the simplest of which is known as "Slater's rules". Note: Zeff is also often written Z*. 3 Values Shielding effect : The shielding effect describes the decrease in attraction between an electron and the nucleus in any atom with more than one electron shell. It is also referred to as the screening effect or atomic shielding. -

Nomenclature of Inorganic Chemistry (IUPAC Recommendations 2005)
NOMENCLATURE OF INORGANIC CHEMISTRY IUPAC Recommendations 2005 IUPAC Periodic Table of the Elements 118 1 2 21314151617 H He 3 4 5 6 7 8 9 10 Li Be B C N O F Ne 11 12 13 14 15 16 17 18 3456 78910 11 12 Na Mg Al Si P S Cl Ar 19 20 21 22 23 24 25 26 27 28 29 30 31 32 33 34 35 36 K Ca Sc Ti V Cr Mn Fe Co Ni Cu Zn Ga Ge As Se Br Kr 37 38 39 40 41 42 43 44 45 46 47 48 49 50 51 52 53 54 Rb Sr Y Zr Nb Mo Tc Ru Rh Pd Ag Cd In Sn Sb Te I Xe 55 56 * 57− 71 72 73 74 75 76 77 78 79 80 81 82 83 84 85 86 Cs Ba lanthanoids Hf Ta W Re Os Ir Pt Au Hg Tl Pb Bi Po At Rn 87 88 ‡ 89− 103 104 105 106 107 108 109 110 111 112 113 114 115 116 117 118 Fr Ra actinoids Rf Db Sg Bh Hs Mt Ds Rg Uub Uut Uuq Uup Uuh Uus Uuo * 57 58 59 60 61 62 63 64 65 66 67 68 69 70 71 La Ce Pr Nd Pm Sm Eu Gd Tb Dy Ho Er Tm Yb Lu ‡ 89 90 91 92 93 94 95 96 97 98 99 100 101 102 103 Ac Th Pa U Np Pu Am Cm Bk Cf Es Fm Md No Lr International Union of Pure and Applied Chemistry Nomenclature of Inorganic Chemistry IUPAC RECOMMENDATIONS 2005 Issued by the Division of Chemical Nomenclature and Structure Representation in collaboration with the Division of Inorganic Chemistry Prepared for publication by Neil G.