Investigation of the Molecular Basis of Three New Disorders of Brain Growth and Development Identified Amongst the Amish
Total Page:16
File Type:pdf, Size:1020Kb

Load more
Recommended publications
-

Reframing Psychiatry for Precision Medicine
Reframing Psychiatry for Precision Medicine Elizabeth B Torres 1,2,3* 1 Rutgers University Department of Psychology; [email protected] 2 Rutgers University Center for Cognitive Science (RUCCS) 3 Rutgers University Computer Science, Center for Biomedicine Imaging and Modelling (CBIM) * Correspondence: [email protected]; Tel.: (011) +858-445-8909 (E.B.T) Supplementary Material Sample Psychological criteria that sidelines sensory motor issues in autism: The ADOS-2 manual [1, 2], under the “Guidelines for Selecting a Module” section states (emphasis added): “Note that the ADOS-2 was developed for and standardized using populations of children and adults without significant sensory and motor impairments. Standardized use of any ADOS-2 module presumes that the individual can walk independently and is free of visual or hearing impairments that could potentially interfere with use of the materials or participation in specific tasks.” Sample Psychiatric criteria from the DSM-5 [3] that does not include sensory-motor issues: A. Persistent deficits in social communication and social interaction across multiple contexts, as manifested by the following, currently or by history (examples are illustrative, not exhaustive, see text): 1. Deficits in social-emotional reciprocity, ranging, for example, from abnormal social approach and failure of normal back-and-forth conversation; to reduced sharing of interests, emotions, or affect; to failure to initiate or respond to social interactions. 2. Deficits in nonverbal communicative behaviors used for social interaction, ranging, for example, from poorly integrated verbal and nonverbal communication; to abnormalities in eye contact and body language or deficits in understanding and use of gestures; to a total lack of facial expressions and nonverbal communication. -

A Computational Approach for Defining a Signature of Β-Cell Golgi Stress in Diabetes Mellitus
Page 1 of 781 Diabetes A Computational Approach for Defining a Signature of β-Cell Golgi Stress in Diabetes Mellitus Robert N. Bone1,6,7, Olufunmilola Oyebamiji2, Sayali Talware2, Sharmila Selvaraj2, Preethi Krishnan3,6, Farooq Syed1,6,7, Huanmei Wu2, Carmella Evans-Molina 1,3,4,5,6,7,8* Departments of 1Pediatrics, 3Medicine, 4Anatomy, Cell Biology & Physiology, 5Biochemistry & Molecular Biology, the 6Center for Diabetes & Metabolic Diseases, and the 7Herman B. Wells Center for Pediatric Research, Indiana University School of Medicine, Indianapolis, IN 46202; 2Department of BioHealth Informatics, Indiana University-Purdue University Indianapolis, Indianapolis, IN, 46202; 8Roudebush VA Medical Center, Indianapolis, IN 46202. *Corresponding Author(s): Carmella Evans-Molina, MD, PhD ([email protected]) Indiana University School of Medicine, 635 Barnhill Drive, MS 2031A, Indianapolis, IN 46202, Telephone: (317) 274-4145, Fax (317) 274-4107 Running Title: Golgi Stress Response in Diabetes Word Count: 4358 Number of Figures: 6 Keywords: Golgi apparatus stress, Islets, β cell, Type 1 diabetes, Type 2 diabetes 1 Diabetes Publish Ahead of Print, published online August 20, 2020 Diabetes Page 2 of 781 ABSTRACT The Golgi apparatus (GA) is an important site of insulin processing and granule maturation, but whether GA organelle dysfunction and GA stress are present in the diabetic β-cell has not been tested. We utilized an informatics-based approach to develop a transcriptional signature of β-cell GA stress using existing RNA sequencing and microarray datasets generated using human islets from donors with diabetes and islets where type 1(T1D) and type 2 diabetes (T2D) had been modeled ex vivo. To narrow our results to GA-specific genes, we applied a filter set of 1,030 genes accepted as GA associated. -

Sensory Deficits in a Mouse Model of Christianson Syndrome Tarheen
Sensory Deficits in a Mouse Model of Christianson Syndrome Tarheen Fatima Department of Physiology McGill University, Montreal April 2019 A thesis submitted to McGill University in partial fulfillment of the requirements of the degree of Master of Science. © Tarheen Fatima 2019 Abstract Christianson Syndrome (CS) is a recently characterized X-linked neurodevelopmental disorder caused by loss-of-function mutations in the gene slc9a6, encoding the endosomal Na+/H+ Exchanger 6 (NHE6). The disorder is associated with developmental delay, intellectual disability, loss of motor coordination, mutism, ataxia, epilepsy, as well as autistic features. In addition to these symptoms, CS patients exhibit elevated pain thresholds to noxious stimuli as well as discomfort at normally innocuous stimuli. The underlying causes of these sensory deficits are yet to be determined. Hence, this study aims at understanding how loss-of-function of NHE6 affects transmission and processing of pain. To this end, we examined the expression of NHE6 in peripheral and central neurons implicated in sensation and interpretation of pain. Additionally, we characterized the nociceptive behaviour of NHE6 knockout (KO) mice using a battery of behaviour tests. Our immunohistochemical experiments demonstrate that NHE6 is highly expressed in nociceptive, small-diameter dorsal root ganglia (DRG) neurons. Moreover, mice lacking NHE6 display decreased responses to noxious mechanical, thermal and chemical stimuli but are more responsive to noxious cold than wild-type littermates. Interestingly, immunohistochemical characterization of DRG tissue from aged NHE6 null mice indicates a decrease in some neuronal subsets suggesting cell death. Finally, using light brush-induced Fos activation in the dorsal horn, we found that the spinal processing of innocuous stimuli is not different between wildtype and NHE6 knockout mice. -

What Is Christianson Syndrome? by Dr
What is Christianson Syndrome? By Dr. Eric Morrow Christianson syndrome is a genetic disorder that affects brain development. It is an “X-linked” disorder (see “Why is CS found only in boys?” section for a description of what X-linked means). Symptoms of CS usually appear in infancy and include epilepsy and intellectual disability. Although the exact prevalence of Christianson syndrome is unknown, a genetic study by Tarpey and colleagues identified Christianson Syndrome in 2 of about 200 families (about 1% of families with apparent X-linked developmental disabilities). This was among the most common genetic change in X-linked forms of intellectual disability. What are the causes of Christianson syndrome? Christianson syndrome is caused by changes in the gene, SLC9A6. Genes are important because they code for proteins, which carry out essential processes in the cells of our bodies. The SLC9A6 gene codes for the NHE6 protein. NHE6 is part of a family of proteins that are sodium (Na+)/hydrogen (H+) exchangers. The NHE6 protein is found in endosomes, which take up molecules to be destroyed or recycled for other processes in our body. NHE6 makes sure that these endosomes have the correct intra-endosomal acid level by letting in Na into the endosome and getting rid of hydrogen, which is the acid molecule. The acid level regulates protein turnover. My analogy our stomach acid, the acidity in endosomes helps degrade cellular proteins. These genetic changes in SLC9A6 cause the NHE6 protein to not work properly. You may have heard words such as “loss-of-function”, “truncation” or “frame- shift”, which describe how the NHE6 protein cannot be made in the cells to carry out these processes in the endosome. -

Luca Bartolini, MD 1 8/9/2021 CURRICULUM VITAE LUCA
8/9/2021 CURRICULUM VITAE LUCA BARTOLINI, MD Director, Pediatric Epilepsy Program Hasbro Children’s Hospital Assistant Professor of Pediatrics Assistant Professor of Neurology Assistant Professor of Neurosurgery The Warren Alpert Medical School of Brown University Email: [email protected] EDUCATION: Undergraduate Liceo Classico “Tito Livio”, Padova, Italy Medical School University of Padova School of Medicine, Padova, Italy 09/2000 – 07/2006 Doctor of Medicine (M.D.) POSTGRADUATE TRAINING: Residency Obstetrics and Gynecology University of Parma, Italy 07/2007 – 01/2008 Pediatrics (Pediatric Neurology Track) University of Padova, Italy 04/2008 – 10/2013 Pediatrics Children’s National Medical Center / The George Washington University, Washington DC 07/2012 – 06/2013 Child Neurology Children’s National Medical Center / The George Washington University, Washington DC 07/2013 – 06/2016 Luca Bartolini, MD 1 Fellowship Neuroscience Research Children’s National Medical Center / The George Washington University, Washington DC National Institutes of Health, Bethesda, MD 07/2016 – 06/2017 Epilepsy National Institutes of Health, Bethesda, MD 07/2017 – 06/2018 Clinical Neurophysiology National Institutes of Health, Bethesda, MD 07/2018 – 06/2019 POSTGRADUATE HONORS AND AWARDS: Research Career Symposium Scholarship American Academy of Neurology 02/2015 Pediatric Epilepsy Fellow Pipeline Travel Scholarship Epilepsy Foundation of America 12/2015 Sentinel of Science Award (Top 10% of Researchers contributing to the peer-review of the field of Medicine) -

Amyloid Clearance Defect in Apoe4 Astrocytes Is Reversed by Epigenetic Correction of Endosomal Ph
Amyloid clearance defect in ApoE4 astrocytes is reversed by epigenetic correction of endosomal pH Hari Prasada and Rajini Raoa,1 aDepartment of Physiology, The Johns Hopkins University School of Medicine, Baltimore, MD 21205 Edited by Reinhard Jahn, Max Planck Institute for Biophysical Chemistry, Goettingen, Germany, and approved June 6, 2018 (received for review January 28, 2018) Endosomes have emerged as a central hub and pathogenic driver ability (7–9). Patients who have CS also show striking age- of Alzheimer’s disease (AD). The earliest brain cytopathology in dependent neurodegeneration, with prominent glial pathology neurodegeneration, occurring decades before amyloid plaques and phosphorylated tau deposits (7). Female carriers have and cognitive decline, is an expansion in the size and number of learning difficulties and behavioral issues, and some present with endosomal compartments. The strongest genetic risk factor for low Mini-Mental State Examination scores suggestive of early sporadic AD is the e4 allele of Apolipoprotein E (ApoE4). Previous cognitive decline (10). Interestingly, NHE6 was among the most studies have shown that ApoE4 potentiates presymptomatic endo- highly down-regulated genes (up to sixfold) in the elderly (70 y) somal dysfunction and defective endocytic clearance of amyloid brain, compared with the adult (40 y) brain (11). These obser- beta (Aβ), although how these two pathways are linked at a cel- vations led us to consider a broader role for NHE6 in neuro- lular and mechanistic level has been unclear. Here, we show that degenerative disorders, including Alzheimer’s disease (AD), a aberrant endosomal acidification in ApoE4 astrocytes traps the major cause of dementia in the elderly. -

Associated 16P11.2 Deletion in Drosophila Melanogaster
ARTICLE DOI: 10.1038/s41467-018-04882-6 OPEN Pervasive genetic interactions modulate neurodevelopmental defects of the autism- associated 16p11.2 deletion in Drosophila melanogaster Janani Iyer1, Mayanglambam Dhruba Singh1, Matthew Jensen1,2, Payal Patel 1, Lucilla Pizzo1, Emily Huber1, Haley Koerselman3, Alexis T. Weiner 1, Paola Lepanto4, Komal Vadodaria1, Alexis Kubina1, Qingyu Wang 1,2, Abigail Talbert1, Sneha Yennawar1, Jose Badano 4, J. Robert Manak3,5, Melissa M. Rolls1, Arjun Krishnan6,7 & 1234567890():,; Santhosh Girirajan 1,2,8 As opposed to syndromic CNVs caused by single genes, extensive phenotypic heterogeneity in variably-expressive CNVs complicates disease gene discovery and functional evaluation. Here, we propose a complex interaction model for pathogenicity of the autism-associated 16p11.2 deletion, where CNV genes interact with each other in conserved pathways to modulate expression of the phenotype. Using multiple quantitative methods in Drosophila RNAi lines, we identify a range of neurodevelopmental phenotypes for knockdown of indi- vidual 16p11.2 homologs in different tissues. We test 565 pairwise knockdowns in the developing eye, and identify 24 interactions between pairs of 16p11.2 homologs and 46 interactions between 16p11.2 homologs and neurodevelopmental genes that suppress or enhance cell proliferation phenotypes compared to one-hit knockdowns. These interac- tions within cell proliferation pathways are also enriched in a human brain-specific network, providing translational relevance in humans. Our study indicates a role for pervasive genetic interactions within CNVs towards cellular and developmental phenotypes. 1 Department of Biochemistry and Molecular Biology, The Pennsylvania State University, University Park, PA 16802, USA. 2 Bioinformatics and Genomics Program, The Huck Institutes of the Life Sciences, The Pennsylvania State University, University Park, PA 16802, USA. -

Supplementary Materials
Supplementary materials Supplementary Table S1: MGNC compound library Ingredien Molecule Caco- Mol ID MW AlogP OB (%) BBB DL FASA- HL t Name Name 2 shengdi MOL012254 campesterol 400.8 7.63 37.58 1.34 0.98 0.7 0.21 20.2 shengdi MOL000519 coniferin 314.4 3.16 31.11 0.42 -0.2 0.3 0.27 74.6 beta- shengdi MOL000359 414.8 8.08 36.91 1.32 0.99 0.8 0.23 20.2 sitosterol pachymic shengdi MOL000289 528.9 6.54 33.63 0.1 -0.6 0.8 0 9.27 acid Poricoic acid shengdi MOL000291 484.7 5.64 30.52 -0.08 -0.9 0.8 0 8.67 B Chrysanthem shengdi MOL004492 585 8.24 38.72 0.51 -1 0.6 0.3 17.5 axanthin 20- shengdi MOL011455 Hexadecano 418.6 1.91 32.7 -0.24 -0.4 0.7 0.29 104 ylingenol huanglian MOL001454 berberine 336.4 3.45 36.86 1.24 0.57 0.8 0.19 6.57 huanglian MOL013352 Obacunone 454.6 2.68 43.29 0.01 -0.4 0.8 0.31 -13 huanglian MOL002894 berberrubine 322.4 3.2 35.74 1.07 0.17 0.7 0.24 6.46 huanglian MOL002897 epiberberine 336.4 3.45 43.09 1.17 0.4 0.8 0.19 6.1 huanglian MOL002903 (R)-Canadine 339.4 3.4 55.37 1.04 0.57 0.8 0.2 6.41 huanglian MOL002904 Berlambine 351.4 2.49 36.68 0.97 0.17 0.8 0.28 7.33 Corchorosid huanglian MOL002907 404.6 1.34 105 -0.91 -1.3 0.8 0.29 6.68 e A_qt Magnogrand huanglian MOL000622 266.4 1.18 63.71 0.02 -0.2 0.2 0.3 3.17 iolide huanglian MOL000762 Palmidin A 510.5 4.52 35.36 -0.38 -1.5 0.7 0.39 33.2 huanglian MOL000785 palmatine 352.4 3.65 64.6 1.33 0.37 0.7 0.13 2.25 huanglian MOL000098 quercetin 302.3 1.5 46.43 0.05 -0.8 0.3 0.38 14.4 huanglian MOL001458 coptisine 320.3 3.25 30.67 1.21 0.32 0.9 0.26 9.33 huanglian MOL002668 Worenine -

Pathway and Gene-Set Activation Measurement from Mrna Expression Data
Open Access Research2006LevineetVolume al. 7, Issue 10, Article R93 Pathway and gene-set activation measurement from mRNA comment expression data: the tissue distribution of human pathways David M Levine*, David R Haynor†, John C Castle*, Sergey B Stepaniants*, Matteo Pellegrini‡, Mao Mao* and Jason M Johnson* Addresses: *Rosetta Inpharmatics LLC, a wholly owned subsidiary of Merck and Co., Inc., Terry Avenue North, Seattle, WA 98109, USA. †Department of Radiology, University of Washington, Seattle, WA 98195, USA. ‡Department of MCD Biology, University of California at Los Angeles, Los Angeles, CA 90095, USA. reviews Correspondence: Jason M Johnson. Email: [email protected] Published: 17 October 2006 Received: 12 July 2006 Revised: 13 September 2006 Genome Biology 2006, 7:R93 (doi:10.1186/gb-2006-7-10-r93) Accepted: 17 October 2006 The electronic version of this article is the complete one and can be found online at http://genomebiology.com/2006/7/10/R93 reports © 2006 Levine et al.; licensee BioMed Central Ltd. This is an open access article distributed under the terms of the Creative Commons Attribution License (http://creativecommons.org/licenses/by/2.0), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited. The<p>Asented.</p> tissue comparison distribution of five of differenthuman pathways measures of pathway expression and a public map of pathway expression in human tissues are pre- deposited research Abstract Background: Interpretation of lists of genes or proteins with altered expression is a critical and time-consuming part of microarray and proteomics research, but relatively little attention has been paid to methods for extracting biological meaning from these output lists. -

Research Resources: Brown University
7/31/2019 This resource listing is intended as a source of information that can be selectively cut, pasted and edited into Facilities and Other Resources sections of grant submissions. BROWN ENVIRONMENT AND RESOURCES (QUICK SECTION LINKS) ACADEMIC INSTITUTIONS ACADEMIC HEALTH CENTERS ACADEMIC SCHOOLS, COLLEGES, DEPARTMENTS, CENTERS, INSTITUTES AND PROGRAMS CLINICAL RESEARCH SUPPORT COLLABORATING INSTITUTIONS COMMUNITY ENGAGEMENT ORGANIZATIONS COMPUTING AND INFORMATION SERVICES CORE AND SHARED RESEARCH FACILITIES DIVERSITY PROGRAMS EDUCATION AND MENTORING RESOURCES EDUCATION PROGRAMS EDUCATION TECHNOLOGY RESOURCES LIBRARY RESOURCES MULTISPECIALTY PRACTICE GROUPS RESEARCH ADMINISTRATION RESEARCH BUILDINGS RHODE ISLAND & RESEARCH ENVIRONMENT RHODE ISLAND STATEWIDE COLLABORATIVE PROGRAMS BROWN ENVIRONMENT AND RESOURCES – DETAILED TABLE OF CONTENTS WITH LINKS ACADEMIC INSTITUTIONS Brown University – Long Brown University - Short University of Rhode Island (URI) ACADEMIC HEALTH CENTERS Lifespan Health System Care New England (CNE) Health System Providence VA Medical Center (PVAMC) ACADEMIC SCHOOLS, COLLEGES, DEPARTMENTS, CENTERS, INSTITUTES AND PROGRAMS Center - Advance-CTR: Comprehensive Center - Advance-CTR: Comprehensive - Alternative Center - Advance-CTR: Summary Center - Advance Predictive Biology Center - Alcohol and Addiction Studies (CAAS) Center - Biology of Aging Center - Brown Center for Biomedical Informatics (BCBI) Center - Brown Center for the Study of Children at Risk Center - Center for Biomedical Engineering (CBME) Center - -
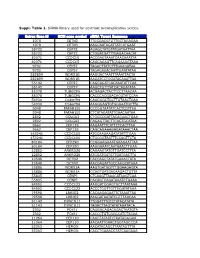
Suppl. Table 1
Suppl. Table 1. SiRNA library used for centriole overduplication screen. Entrez Gene Id NCBI gene symbol siRNA Target Sequence 1070 CETN3 TTGCGACGTGTTGCTAGAGAA 1070 CETN3 AAGCAATAGATTATCATGAAT 55722 CEP72 AGAGCTATGTATGATAATTAA 55722 CEP72 CTGGATGATTTGAGACAACAT 80071 CCDC15 ACCGAGTAAATCAACAAATTA 80071 CCDC15 CAGCAGAGTTCAGAAAGTAAA 9702 CEP57 TAGACTTATCTTTGAAGATAA 9702 CEP57 TAGAGAAACAATTGAATATAA 282809 WDR51B AAGGACTAATTTAAATTACTA 282809 WDR51B AAGATCCTGGATACAAATTAA 55142 CEP27 CAGCAGATCACAAATATTCAA 55142 CEP27 AAGCTGTTTATCACAGATATA 85378 TUBGCP6 ACGAGACTACTTCCTTAACAA 85378 TUBGCP6 CACCCACGGACACGTATCCAA 54930 C14orf94 CAGCGGCTGCTTGTAACTGAA 54930 C14orf94 AAGGGAGTGTGGAAATGCTTA 5048 PAFAH1B1 CCCGGTAATATCACTCGTTAA 5048 PAFAH1B1 CTCATAGATATTGAACAATAA 2802 GOLGA3 CTGGCCGATTACAGAACTGAA 2802 GOLGA3 CAGAGTTACTTCAGTGCATAA 9662 CEP135 AAGAATTTCATTCTCACTTAA 9662 CEP135 CAGCAGAAAGAGATAAACTAA 153241 CCDC100 ATGCAAGAAGATATATTTGAA 153241 CCDC100 CTGCGGTAATTTCCAGTTCTA 80184 CEP290 CCGGAAGAAATGAAGAATTAA 80184 CEP290 AAGGAAATCAATAAACTTGAA 22852 ANKRD26 CAGAAGTATGTTGATCCTTTA 22852 ANKRD26 ATGGATGATGTTGATGACTTA 10540 DCTN2 CACCAGCTATATGAAACTATA 10540 DCTN2 AACGAGATTGCCAAGCATAAA 25886 WDR51A AAGTGATGGTTTGGAAGAGTA 25886 WDR51A CCAGTGATGACAAGACTGTTA 55835 CENPJ CTCAAGTTAAACATAAGTCAA 55835 CENPJ CACAGTCAGATAAATCTGAAA 84902 CCDC123 AAGGATGGAGTGCTTAATAAA 84902 CCDC123 ACCCTGGTTGTTGGATATAAA 79598 LRRIQ2 CACAAGAGAATTCTAAATTAA 79598 LRRIQ2 AAGGATAATATCGTTTAACAA 51143 DYNC1LI1 TTGGATTTGTCTATACATATA 51143 DYNC1LI1 TAGACTTAGTATATAAATACA 2302 FOXJ1 CAGGACAGACAGACTAATGTA -

A Potential Gain-Of-Function Variant of SLC9A6 Leads to Endosomal T Alkalinization and Neuronal Atrophy Associated with Christianson Syndrome Alina Iliea, Andy Y.L
Neurobiology of Disease 121 (2019) 187–204 Contents lists available at ScienceDirect Neurobiology of Disease journal homepage: www.elsevier.com/locate/ynbdi A potential gain-of-function variant of SLC9A6 leads to endosomal T alkalinization and neuronal atrophy associated with Christianson Syndrome Alina Iliea, Andy Y.L. Gaob, Annie Bouchera, Jaeok Parkc, Albert M. Berghuisc, ⁎ Mariëtte J.V. Hofferd, Yvonne Hilhorst-Hofsteed, R. Anne McKinneyb, John Orlowskia, a Department of Physiology, McGill University, Montreal, Canada b Department of Pharmacology and Therapeutics, McGill University, Montreal, Canada c Department of Biochemistry, McGill University, Montreal, Canada d Department of Clinical Genetics, Leiden University Medical Center, Leiden, the Netherlands ARTICLE INFO ABSTRACT Keywords: Loss-of-function mutations in the recycling endosomal (Na+,K+)/H+ exchanger gene SLC9A6/NHE6 result in Christianson Syndrome overacidification and dysfunction of endosomal-lysosomal compartments, and cause a neurodevelopmental and X-linked intellectual disability degenerative form of X-linked intellectual disability called Christianson Syndrome (CS). However, knowledge of Alkali cation/proton exchangers –SLC9A6/ the disease heterogeneity of CS is limited. Here, we describe the clinical features and underlying molecular and NHE6 cellular mechanisms associated with a CS patient carrying a de novo missense variant (p.Gly218Arg; G218R) of a Endosomal pH homeostasis conserved residue in its ion translocation domain that results in a potential gain-of-function. The patient Membrane trafficking Neurodegeneration manifested several core symptoms typical of CS, including pronounced cognitive impairment, mutism, epilepsy, ataxia and microcephaly; however, deterioration of motor function often observed after the first decade of life in CS children with total loss of SLC9A6/NHE6 function was not evident.