Pathway and Gene-Set Activation Measurement from Mrna Expression Data
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

The Heterotaxy Candidate Gene, TMEM195, Regulates Nuclear Localization of Beta-Catenin
Yale University EliScholar – A Digital Platform for Scholarly Publishing at Yale Yale Medicine Thesis Digital Library School of Medicine January 2015 The etH erotaxy Candidate Gene, Tmem195, Regulates Nuclear Localization Of Beta-Catenin Anna Ruth Duncan Yale School of Medicine, [email protected] Follow this and additional works at: http://elischolar.library.yale.edu/ymtdl Recommended Citation Duncan, Anna Ruth, "The eH terotaxy Candidate Gene, Tmem195, Regulates Nuclear Localization Of Beta-Catenin" (2015). Yale Medicine Thesis Digital Library. 1961. http://elischolar.library.yale.edu/ymtdl/1961 This Open Access Thesis is brought to you for free and open access by the School of Medicine at EliScholar – A Digital Platform for Scholarly Publishing at Yale. It has been accepted for inclusion in Yale Medicine Thesis Digital Library by an authorized administrator of EliScholar – A Digital Platform for Scholarly Publishing at Yale. For more information, please contact [email protected]. The Heterotaxy Candidate Gene, TMEM195, Regulates Nuclear Localization of Beta-catenin A Thesis Submitted to the Yale University School of Medicine in Partial Fulfillment of the Requirements for the Degree of Doctor of Medicine By Anna Ruth Duncan 2015 Abstract THE HETEROTAXY CANDIDATE GENE, TMEM195, REGULATES NUCLEAR LOCALIZATION OF BETA-CATENIN. Anna R. Duncan, John Griffin, Andrew Robson, and Mustafa K. Khokha. Department of Pediatrics, Yale University, School of Medicine, New Haven, CT. Congenital heart disease (CHD) affects 1 in every 130 newborns and is the leading cause of infant mortality (2). Heterotaxy (Htx), a disorder of left-right (LR) development, commonly leads to CHD. Despite aggressive surgical management, patients with Htx have poor survival rates and severe morbidity due to their complex CHD. -

A Computational Approach for Defining a Signature of Β-Cell Golgi Stress in Diabetes Mellitus
Page 1 of 781 Diabetes A Computational Approach for Defining a Signature of β-Cell Golgi Stress in Diabetes Mellitus Robert N. Bone1,6,7, Olufunmilola Oyebamiji2, Sayali Talware2, Sharmila Selvaraj2, Preethi Krishnan3,6, Farooq Syed1,6,7, Huanmei Wu2, Carmella Evans-Molina 1,3,4,5,6,7,8* Departments of 1Pediatrics, 3Medicine, 4Anatomy, Cell Biology & Physiology, 5Biochemistry & Molecular Biology, the 6Center for Diabetes & Metabolic Diseases, and the 7Herman B. Wells Center for Pediatric Research, Indiana University School of Medicine, Indianapolis, IN 46202; 2Department of BioHealth Informatics, Indiana University-Purdue University Indianapolis, Indianapolis, IN, 46202; 8Roudebush VA Medical Center, Indianapolis, IN 46202. *Corresponding Author(s): Carmella Evans-Molina, MD, PhD ([email protected]) Indiana University School of Medicine, 635 Barnhill Drive, MS 2031A, Indianapolis, IN 46202, Telephone: (317) 274-4145, Fax (317) 274-4107 Running Title: Golgi Stress Response in Diabetes Word Count: 4358 Number of Figures: 6 Keywords: Golgi apparatus stress, Islets, β cell, Type 1 diabetes, Type 2 diabetes 1 Diabetes Publish Ahead of Print, published online August 20, 2020 Diabetes Page 2 of 781 ABSTRACT The Golgi apparatus (GA) is an important site of insulin processing and granule maturation, but whether GA organelle dysfunction and GA stress are present in the diabetic β-cell has not been tested. We utilized an informatics-based approach to develop a transcriptional signature of β-cell GA stress using existing RNA sequencing and microarray datasets generated using human islets from donors with diabetes and islets where type 1(T1D) and type 2 diabetes (T2D) had been modeled ex vivo. To narrow our results to GA-specific genes, we applied a filter set of 1,030 genes accepted as GA associated. -

Associated 16P11.2 Deletion in Drosophila Melanogaster
ARTICLE DOI: 10.1038/s41467-018-04882-6 OPEN Pervasive genetic interactions modulate neurodevelopmental defects of the autism- associated 16p11.2 deletion in Drosophila melanogaster Janani Iyer1, Mayanglambam Dhruba Singh1, Matthew Jensen1,2, Payal Patel 1, Lucilla Pizzo1, Emily Huber1, Haley Koerselman3, Alexis T. Weiner 1, Paola Lepanto4, Komal Vadodaria1, Alexis Kubina1, Qingyu Wang 1,2, Abigail Talbert1, Sneha Yennawar1, Jose Badano 4, J. Robert Manak3,5, Melissa M. Rolls1, Arjun Krishnan6,7 & 1234567890():,; Santhosh Girirajan 1,2,8 As opposed to syndromic CNVs caused by single genes, extensive phenotypic heterogeneity in variably-expressive CNVs complicates disease gene discovery and functional evaluation. Here, we propose a complex interaction model for pathogenicity of the autism-associated 16p11.2 deletion, where CNV genes interact with each other in conserved pathways to modulate expression of the phenotype. Using multiple quantitative methods in Drosophila RNAi lines, we identify a range of neurodevelopmental phenotypes for knockdown of indi- vidual 16p11.2 homologs in different tissues. We test 565 pairwise knockdowns in the developing eye, and identify 24 interactions between pairs of 16p11.2 homologs and 46 interactions between 16p11.2 homologs and neurodevelopmental genes that suppress or enhance cell proliferation phenotypes compared to one-hit knockdowns. These interac- tions within cell proliferation pathways are also enriched in a human brain-specific network, providing translational relevance in humans. Our study indicates a role for pervasive genetic interactions within CNVs towards cellular and developmental phenotypes. 1 Department of Biochemistry and Molecular Biology, The Pennsylvania State University, University Park, PA 16802, USA. 2 Bioinformatics and Genomics Program, The Huck Institutes of the Life Sciences, The Pennsylvania State University, University Park, PA 16802, USA. -

Supplementary Materials
Supplementary materials Supplementary Table S1: MGNC compound library Ingredien Molecule Caco- Mol ID MW AlogP OB (%) BBB DL FASA- HL t Name Name 2 shengdi MOL012254 campesterol 400.8 7.63 37.58 1.34 0.98 0.7 0.21 20.2 shengdi MOL000519 coniferin 314.4 3.16 31.11 0.42 -0.2 0.3 0.27 74.6 beta- shengdi MOL000359 414.8 8.08 36.91 1.32 0.99 0.8 0.23 20.2 sitosterol pachymic shengdi MOL000289 528.9 6.54 33.63 0.1 -0.6 0.8 0 9.27 acid Poricoic acid shengdi MOL000291 484.7 5.64 30.52 -0.08 -0.9 0.8 0 8.67 B Chrysanthem shengdi MOL004492 585 8.24 38.72 0.51 -1 0.6 0.3 17.5 axanthin 20- shengdi MOL011455 Hexadecano 418.6 1.91 32.7 -0.24 -0.4 0.7 0.29 104 ylingenol huanglian MOL001454 berberine 336.4 3.45 36.86 1.24 0.57 0.8 0.19 6.57 huanglian MOL013352 Obacunone 454.6 2.68 43.29 0.01 -0.4 0.8 0.31 -13 huanglian MOL002894 berberrubine 322.4 3.2 35.74 1.07 0.17 0.7 0.24 6.46 huanglian MOL002897 epiberberine 336.4 3.45 43.09 1.17 0.4 0.8 0.19 6.1 huanglian MOL002903 (R)-Canadine 339.4 3.4 55.37 1.04 0.57 0.8 0.2 6.41 huanglian MOL002904 Berlambine 351.4 2.49 36.68 0.97 0.17 0.8 0.28 7.33 Corchorosid huanglian MOL002907 404.6 1.34 105 -0.91 -1.3 0.8 0.29 6.68 e A_qt Magnogrand huanglian MOL000622 266.4 1.18 63.71 0.02 -0.2 0.2 0.3 3.17 iolide huanglian MOL000762 Palmidin A 510.5 4.52 35.36 -0.38 -1.5 0.7 0.39 33.2 huanglian MOL000785 palmatine 352.4 3.65 64.6 1.33 0.37 0.7 0.13 2.25 huanglian MOL000098 quercetin 302.3 1.5 46.43 0.05 -0.8 0.3 0.38 14.4 huanglian MOL001458 coptisine 320.3 3.25 30.67 1.21 0.32 0.9 0.26 9.33 huanglian MOL002668 Worenine -
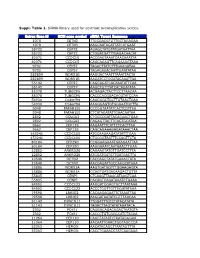
Suppl. Table 1
Suppl. Table 1. SiRNA library used for centriole overduplication screen. Entrez Gene Id NCBI gene symbol siRNA Target Sequence 1070 CETN3 TTGCGACGTGTTGCTAGAGAA 1070 CETN3 AAGCAATAGATTATCATGAAT 55722 CEP72 AGAGCTATGTATGATAATTAA 55722 CEP72 CTGGATGATTTGAGACAACAT 80071 CCDC15 ACCGAGTAAATCAACAAATTA 80071 CCDC15 CAGCAGAGTTCAGAAAGTAAA 9702 CEP57 TAGACTTATCTTTGAAGATAA 9702 CEP57 TAGAGAAACAATTGAATATAA 282809 WDR51B AAGGACTAATTTAAATTACTA 282809 WDR51B AAGATCCTGGATACAAATTAA 55142 CEP27 CAGCAGATCACAAATATTCAA 55142 CEP27 AAGCTGTTTATCACAGATATA 85378 TUBGCP6 ACGAGACTACTTCCTTAACAA 85378 TUBGCP6 CACCCACGGACACGTATCCAA 54930 C14orf94 CAGCGGCTGCTTGTAACTGAA 54930 C14orf94 AAGGGAGTGTGGAAATGCTTA 5048 PAFAH1B1 CCCGGTAATATCACTCGTTAA 5048 PAFAH1B1 CTCATAGATATTGAACAATAA 2802 GOLGA3 CTGGCCGATTACAGAACTGAA 2802 GOLGA3 CAGAGTTACTTCAGTGCATAA 9662 CEP135 AAGAATTTCATTCTCACTTAA 9662 CEP135 CAGCAGAAAGAGATAAACTAA 153241 CCDC100 ATGCAAGAAGATATATTTGAA 153241 CCDC100 CTGCGGTAATTTCCAGTTCTA 80184 CEP290 CCGGAAGAAATGAAGAATTAA 80184 CEP290 AAGGAAATCAATAAACTTGAA 22852 ANKRD26 CAGAAGTATGTTGATCCTTTA 22852 ANKRD26 ATGGATGATGTTGATGACTTA 10540 DCTN2 CACCAGCTATATGAAACTATA 10540 DCTN2 AACGAGATTGCCAAGCATAAA 25886 WDR51A AAGTGATGGTTTGGAAGAGTA 25886 WDR51A CCAGTGATGACAAGACTGTTA 55835 CENPJ CTCAAGTTAAACATAAGTCAA 55835 CENPJ CACAGTCAGATAAATCTGAAA 84902 CCDC123 AAGGATGGAGTGCTTAATAAA 84902 CCDC123 ACCCTGGTTGTTGGATATAAA 79598 LRRIQ2 CACAAGAGAATTCTAAATTAA 79598 LRRIQ2 AAGGATAATATCGTTTAACAA 51143 DYNC1LI1 TTGGATTTGTCTATACATATA 51143 DYNC1LI1 TAGACTTAGTATATAAATACA 2302 FOXJ1 CAGGACAGACAGACTAATGTA -

Novel Gene Discovery in Primary Ciliary Dyskinesia
Novel Gene Discovery in Primary Ciliary Dyskinesia Mahmoud Raafat Fassad Genetics and Genomic Medicine Programme Great Ormond Street Institute of Child Health University College London A thesis submitted in conformity with the requirements for the degree of Doctor of Philosophy University College London 1 Declaration I, Mahmoud Raafat Fassad, confirm that the work presented in this thesis is my own. Where information has been derived from other sources, I confirm that this has been indicated in the thesis. 2 Abstract Primary Ciliary Dyskinesia (PCD) is one of the ‘ciliopathies’, genetic disorders affecting either cilia structure or function. PCD is a rare recessive disease caused by defective motile cilia. Affected individuals manifest with neonatal respiratory distress, chronic wet cough, upper respiratory tract problems, progressive lung disease resulting in bronchiectasis, laterality problems including heart defects and adult infertility. Early diagnosis and management are essential for better respiratory disease prognosis. PCD is a highly genetically heterogeneous disorder with causal mutations identified in 36 genes that account for the disease in about 70% of PCD cases, suggesting that additional genes remain to be discovered. Targeted next generation sequencing was used for genetic screening of a cohort of patients with confirmed or suggestive PCD diagnosis. The use of multi-gene panel sequencing yielded a high diagnostic output (> 70%) with mutations identified in known PCD genes. Over half of these mutations were novel alleles, expanding the mutation spectrum in PCD genes. The inclusion of patients from various ethnic backgrounds revealed a striking impact of ethnicity on the composition of disease alleles uncovering a significant genetic stratification of PCD in different populations. -

Identification, Tissue Specific Expression, and Chromosomal
European Journal of Human Genetics (2000) 8, 923–932 © 2000 Macmillan Publishers Ltd All rights reserved 1018–4813/00 $15.00 y www.nature.com/ejhg ARTICLE Identification, tissue specific expression, and chromosomal localisation of several human dynein heavy chain genes Amit K Maiti1, Marie-Genevi`eve Matt´ei2, Mark Jorissen3, Armin Volz4, Andreas Zeigler4 and Patrice Bouvagnet5 1Division of Medical Genetics, Geneva University School of Medicine, Geneva, Switzerland; 2INSERM U 406, Facult´e de M´edecine, Marseille, France; 3Centrum voor Menselijke Erfelijkheid, Campus Gasthuisberg, O & N, Leuven, Belgium; 4Institut f¨ur Immungenetik, Universit¨atsklinikum Charit´e, Humboldt-Universit¨at zu Berlin, Berlin, Germany; 5Laboratoire de G´en´etique Mol´eculaire Humaine, Facult´e de Pharmacie, Lyon, France Sliding between adjacent microtubules within the axonema gives rise to the motility of cilia and flagella. The driving force is produced by dynein complexes which are mainly composed of the axonemal dynein heavy chains. We used cells of human respiratory epithelium after in vitro ciliogenesis to clone cDNA fragments of nine dynein heavy chain genes, one of which had never been identified before. Dynein heavy chains are highly conserved from protozoa to human and the evolutionary ancestry of these dynein heavy chain cDNA fragments was deduced by phylogenetic analysis. These dynein heavy chain cDNAs are highly transcribed in human tissues containing axonema such as trachea, testis and brain, but not in adult heart or placenta. PAC clones containing dynein heavy chains were obtained and used to determine by FISH their chromosomal position in the human genome. They were mapped to 2p12–p11, 2q33, 3p21.2–p21.1, 13q14, 16p12 and 17p12. -

Cardiovascular Diseases Genetic Testing Program Information
Cardiovascular Diseases Genetic Testing Program Description: Congenital Heart Disease Panels We offer comprehensive gene panels designed to • Congenital Heart Disease Panel (187 genes) diagnose the most common genetic causes of hereditary • Heterotaxy Panel (114 genes) cardiovascular diseases. Testing is available for congenital • RASopathy/Noonan Spectrum Disorders Panel heart malformation, cardiomyopathy, arrythmia, thoracic (31 genes) aortic aneurysm, pulmonary arterial hypertension, Marfan Other Panels syndrome, and RASopathy/Noonan spectrum disorders. • Pulmonary Arterial Hypertension (PAH) Panel Hereditary cardiovascular disease is caused by variants in (20 genes) many different genes, and may be inherited in an autosomal dominant, autosomal recessive, or X-linked manner. Other Indications: than condition-specific panels, we also offer single gene Panels: sequencing for any gene on the panels, targeted variant • Confirmation of genetic diagnosis in a patient with analysis, and targeted deletion/duplication analysis. a clinical diagnosis of cardiovascular disease Tests Offered: • Carrier or pre-symptomatic diagnosis identification Arrythmia Panels in individuals with a family history of cardiovascular • Comprehensive Arrhythmia Panel (81 genes) disease of unknown genetic basis • Atrial Fibrillation (A Fib) Panel (28 genes) Gene Specific Sequencing: • Atrioventricular Block (AV Block) Panel (7 genes) • Confirmation of genetic diagnosis in a patient with • Brugada Syndrome Panel (21 genes) cardiovascular disease and in whom a specific -

Genomic Landscape of Paediatric Adrenocortical Tumours
ARTICLE Received 5 Aug 2014 | Accepted 16 Jan 2015 | Published 6 Mar 2015 DOI: 10.1038/ncomms7302 Genomic landscape of paediatric adrenocortical tumours Emilia M. Pinto1,*, Xiang Chen2,*, John Easton2, David Finkelstein2, Zhifa Liu3, Stanley Pounds3, Carlos Rodriguez-Galindo4, Troy C. Lund5, Elaine R. Mardis6,7,8, Richard K. Wilson6,7,9, Kristy Boggs2, Donald Yergeau2, Jinjun Cheng2, Heather L. Mulder2, Jayanthi Manne2, Jesse Jenkins10, Maria J. Mastellaro11, Bonald C. Figueiredo12, Michael A. Dyer13, Alberto Pappo14, Jinghui Zhang2, James R. Downing10, Raul C. Ribeiro14,* & Gerard P. Zambetti1,* Paediatric adrenocortical carcinoma is a rare malignancy with poor prognosis. Here we analyse 37 adrenocortical tumours (ACTs) by whole-genome, whole-exome and/or transcriptome sequencing. Most cases (91%) show loss of heterozygosity (LOH) of chromosome 11p, with uniform selection against the maternal chromosome. IGF2 on chromosome 11p is overexpressed in 100% of the tumours. TP53 mutations and chromosome 17 LOH with selection against wild-type TP53 are observed in 28 ACTs (76%). Chromosomes 11p and 17 undergo copy-neutral LOH early during tumorigenesis, suggesting tumour-driver events. Additional genetic alterations include recurrent somatic mutations in ATRX and CTNNB1 and integration of human herpesvirus-6 in chromosome 11p. A dismal outcome is predicted by concomitant TP53 and ATRX mutations and associated genomic abnormalities, including massive structural variations and frequent background mutations. Collectively, these findings demonstrate the nature, timing and potential prognostic significance of key genetic alterations in paediatric ACT and outline a hypothetical model of paediatric adrenocortical tumorigenesis. 1 Department of Biochemistry, St Jude Children’s Research Hospital, Memphis, Tennessee 38105, USA. 2 Department of Computational Biology and Bioinformatics, St Jude Children’s Research Hospital, Memphis, Tennessee 38105, USA. -

Genes of the Pig, Sus Scrofa, Reconstructed with Evidentialgene
bioRxiv preprint doi: https://doi.org/10.1101/412130; this version posted December 28, 2018. The copyright holder for this preprint (which was not certified by peer review) is the author/funder, who has granted bioRxiv a license to display the preprint in perpetuity. It is made available under aCC-BY-NC-ND 4.0 International license. Pig Genes Reconstructed with EvidentialGene 2018 July Genes of the Pig, Sus scrofa, reconstructed with Evi- dentialGene Author: Donald G. Gilbert Affiliation: Indiana University, Bloomington, IN, USA Email address: [email protected] or [email protected] Abstract The pig is a well-studied model animal of biomedical and agricultural importance. Genes of this species, Sus scrofa, are known from experiments and predictions, and collected at the NCBI Ref- erence Sequence database section. Gene reconstruction from transcribed gene evidence of RNA- seq now can accurately and completely reproduce the biological gene sets of animals and plants. Such a gene set for the pig is reported here, including human orthologs missing from current NCBI and Ensembl reference pig gene sets, additional alternate transcripts, and other improve- ments. Methodology for accurate and complete gene set reconstruction from RNA is used: the automated SRA2Genes pipeline of EvidentialGene project. !1 bioRxiv preprint doi: https://doi.org/10.1101/412130; this version posted December 28, 2018. The copyright holder for this preprint (which was not certified by peer review) is the author/funder, who has granted bioRxiv a license to display the preprint in perpetuity. It is made available under aCC-BY-NC-ND 4.0 International license. -

Research Article Complex and Multidimensional Lipid Raft Alterations in a Murine Model of Alzheimer’S Disease
SAGE-Hindawi Access to Research International Journal of Alzheimer’s Disease Volume 2010, Article ID 604792, 56 pages doi:10.4061/2010/604792 Research Article Complex and Multidimensional Lipid Raft Alterations in a Murine Model of Alzheimer’s Disease Wayne Chadwick, 1 Randall Brenneman,1, 2 Bronwen Martin,3 and Stuart Maudsley1 1 Receptor Pharmacology Unit, National Institute on Aging, National Institutes of Health, 251 Bayview Boulevard, Suite 100, Baltimore, MD 21224, USA 2 Miller School of Medicine, University of Miami, Miami, FL 33124, USA 3 Metabolism Unit, National Institute on Aging, National Institutes of Health, 251 Bayview Boulevard, Suite 100, Baltimore, MD 21224, USA Correspondence should be addressed to Stuart Maudsley, [email protected] Received 17 May 2010; Accepted 27 July 2010 Academic Editor: Gemma Casadesus Copyright © 2010 Wayne Chadwick et al. This is an open access article distributed under the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited. Various animal models of Alzheimer’s disease (AD) have been created to assist our appreciation of AD pathophysiology, as well as aid development of novel therapeutic strategies. Despite the discovery of mutated proteins that predict the development of AD, there are likely to be many other proteins also involved in this disorder. Complex physiological processes are mediated by coherent interactions of clusters of functionally related proteins. Synaptic dysfunction is one of the hallmarks of AD. Synaptic proteins are organized into multiprotein complexes in high-density membrane structures, known as lipid rafts. These microdomains enable coherent clustering of synergistic signaling proteins. -

UNDERSTANDING the ROLE of SPLICING FACTORS in CENTRIOLE DUPLICATION by Elizabeth Michelle Park a Dissertation Submitted to John
UNDERSTANDING THE ROLE OF SPLICING FACTORS IN CENTRIOLE DUPLICATION by Elizabeth Michelle Park A dissertation submitted to Johns Hopkins University in conformity with the requirements for the degree of Doctor of Philosophy Baltimore, Maryland January 2020 © 2020 Elizabeth Park All rights reserved Abstract The centriole is a microtubule-based structure that forms the core of the centrosome, the major microtubule organizing center of the cell. Each centrosome contains precisely two centrioles that are surrounded by pericentriolar material that nucleates microtubules and plays crucial roles in the cell. Cycling cells undergo exactly one round of centriole duplication that is regulated numerically, spatially, and temporally alongside the duplication of the cell’s DNA. This regulation is important for cell health and viability, as aberrations in centriole number can lead to diseases such as cancer. The complete molecular mechanisms regulating centriole duplication have not yet been discovered. Recent work identified that a subset of splicing factors is required for centriole biogenesis. This revealed a previously unstudied means of post-transcriptional regulation that centriole proteins could undergo to ensure that the centriolar building blocks are translated in stoichiometrically appropriate amounts. While this subset of splicing factors was identified as playing a role in centriole duplication, the precise mechanism by which they regulate centriole biogenesis at the post transcriptional level was not identified. I sought to identify novel means of regulating centriole duplication. I uncovered an additional splicing factor, WBP11, that upon depletion, yields similar phenotypes to the depletion of the fourteen other splicing factors implicated in centriole biogenesis. I found that WBP11, SNW1, and likely other centriole-related splicing factors are required to splice out short, weak introns such as those found at the 3’ terminus of the TUBGCP6 pre-mRNA.