Isotopic Fractionation of Barium in Shale and Produced Water from the Appalachian Basin, Usa
Total Page:16
File Type:pdf, Size:1020Kb

Load more
Recommended publications
-

Isotopic Analyses of Barium in Meteorites and in Terrestrial
•'OO•,NALO• GEO•'IIYSlCALRESXAaCX VOL. ?4, NO. IS, JULY IS, 1969 Isotopic Analysesof Barium in Meteorites and in Terrestrial Samples O. EUGSTER,F. TERA,AND G. J. WASSERBURG Charles Arms Laboratory o• the Division o• Geological Sciences Cali•Jrnia Institute o• Technology,Pasadena, California 91109 Isotopic compos•tien and concentration of barium in six stone meteorites and the silicate inclusionsof two iron meteorites and three terrestrial sampleswere measured by use of a 'double spike' isotopic dilution technique in order to correct for laboratory fractionation. Any differencesbetween the abundancesof the isotopesin meteoritic and terrestrial Ba were found to be less than 0.1% for all isotopes.The per cent abundancesof Ba found in our work for Ba '•s, Ba•, Ba•% Ba '•, Ba TM,Ba •', and Ba'•ø are 71.699, 11.232, 7.853, 6.592, 2.417, 0.1012, and 0.1058, respectively. Because of the higher precision, these abundancesshould replace the currently accepted values. These results show the variations in the Ba isotopesreported by S. Umemoto (1962) to be unsubstantiated. INTRODUCTION Umemoto [1962] reported that the isotopic A comparison.of the isotopiccomposition of compositionof Ba in the Bruderheimchondrite and in the Pasamonte and Nuevo Laredo various elements in terrestrial samples,me- achondrites were distinct from terrestrial Ba teorites, and other materials of the solar system is of fundamental importance in determin- and showeda pattern of uniform fractionation relative to terrestrial Ba. The enrichments ob- ing the early history of the solar system and the mechanismsof nucleosynthesis.In ad- servedby him correspondto a 2% enrichment dition to effectsthat are a product of either in the ratio Ba•ø/Ba• compared with ter- long-lived natural activity or cosmic-rayinter- restrial materials. -

6. Potential for Human Exposure
CESIUM 125 6. POTENTIAL FOR HUMAN EXPOSURE 6.1 OVERVIEW Cesium has been identified in at least 8 of the 1,636 hazardous waste sites that have been proposed for inclusion on the EPA National Priorities List (NPL) (HazDat 2003). It was reported that 134Cs has been found in at least 3 of the 1,636 current or former NPL sites and 137Cs has been detected in at least 23 of the 1,636 current or former NPL sites. However, the number of sites evaluated for cesium is not known. The frequency of these sites can be seen in Figures 6-1, 6-2, and 6-3. Of these sites, none are located in the Commonwealth of Puerto Rico. Naturally-occurring cesium and cesium minerals consist of only one stable isotope, 133Cs. Cesium occurs in the earth's crust at low concentrations. Granites contain an average cesium concentration of about 1 ppm and sedimentary rocks contain about 4 ppm (Burt 1993). Higher concentrations are found in lepidolite, carnallite, muscovite, beryl, spodumene, potassium feldspars, leucite, petalite, and related minerals. The most important source of commercial cesium is the mineral pollucite, which usually contains about 5–32% Cs2O (Burt 1993). The largest deposits of pollucite are located in Manitoba, Canada and account for about two-thirds of the world’s known supply. Smaller deposits are located in Zimbabwe, Namibia, Brazil, Scandinavia, Czechoslovakia, and the United States. Continental dust and soil erosion are the main emission sources of naturally occurring cesium present in the environment. Cesium is also released to the environment as a result of human activities. -

The New Nuclear Forensics: Analysis of Nuclear Material for Security
THE NEW NUCLEAR FORENSICS Analysis of Nuclear Materials for Security Purposes edited by vitaly fedchenko The New Nuclear Forensics Analysis of Nuclear Materials for Security Purposes STOCKHOLM INTERNATIONAL PEACE RESEARCH INSTITUTE SIPRI is an independent international institute dedicated to research into conflict, armaments, arms control and disarmament. Established in 1966, SIPRI provides data, analysis and recommendations, based on open sources, to policymakers, researchers, media and the interested public. The Governing Board is not responsible for the views expressed in the publications of the Institute. GOVERNING BOARD Sven-Olof Petersson, Chairman (Sweden) Dr Dewi Fortuna Anwar (Indonesia) Dr Vladimir Baranovsky (Russia) Ambassador Lakhdar Brahimi (Algeria) Jayantha Dhanapala (Sri Lanka) Ambassador Wolfgang Ischinger (Germany) Professor Mary Kaldor (United Kingdom) The Director DIRECTOR Dr Ian Anthony (United Kingdom) Signalistgatan 9 SE-169 70 Solna, Sweden Telephone: +46 8 655 97 00 Fax: +46 8 655 97 33 Email: [email protected] Internet: www.sipri.org The New Nuclear Forensics Analysis of Nuclear Materials for Security Purposes EDITED BY VITALY FEDCHENKO OXFORD UNIVERSITY PRESS 2015 1 Great Clarendon Street, Oxford OX2 6DP, United Kingdom Oxford University Press is a department of the University of Oxford. It furthers the University’s objective of excellence in research, scholarship, and education by publishing worldwide. Oxford is a registered trade mark of Oxford University Press in the UK and in certain other countries © SIPRI 2015 The moral rights of the authors have been asserted All rights reserved. No part of this publication may be reproduced, stored in a retrieval system, or transmitted, in any form or by any means, without the prior permission in writing of SIPRI, or as expressly permitted by law, or under terms agreed with the appropriate reprographics rights organizations. -

Atoms Worksheet #2 (The Mole)
Name_______________________________ Atoms Worksheet #2 (The Mole) 1. Counting Atoms – How many atoms are in the following compounds? a) CaCl2___3____ b) NH4OH___7____ c) NaCl____2___ d) N2O7__9_____ e) P2O5__7_____ f) KNO3___5____ g) Al2(CO3)3__14_____ h) Na3PO4__8___ i) Mg(NO3)2__9_____ 2. Calculate the molar mass of the following compounds: a) NaCl 58.5 g/mol b) Fe2O3 159.6 g/mol c) LiOH 23.9 g/mol d) NH3 17.0 g/mol e) AlPO4 122.3 g/mol 3. Mole-to-Gram/Gram-to-Mole Conversions (One Step Problems) a) How many grams of Ca are present in 3.28 moles? 132 g Ca b) How many grams of S are present in 5.39 moles? 173 g S c) How many moles of Ag are there in 4.98 gram? 0.0462 mol d) How many moles of Mg are present in 303 grams? 12.5 mol Mg 4. Conversions using Avogadro’s number (One Step Problems) a) How many atoms are present in 34.69 moles of Mg? 2.09 X 1025 atoms b) How many atoms are present in 0.529 moles of Li? 3.18 X 1023 atoms c) How many moles of Mn are present in 4.09 x 1024 atoms of Mn? 6.79 mol d) How many moles of Ni are present in 5.88 x 1019 atoms? 0.0000977 mol 5. Grams ↔ Moles ↔ Particles OR Atoms OR Molecules (Two Step Problems) a) Calculate the number of molecules in 8.33 grams of O2 1.57 X 1023 molecules b.) Calculate the number of atoms in 43.33 grams of iron, Fe? 4.67 X 1023 atoms c) Calculate the number of particles in 32.8 grams of Cu2S. -

Fingerprints of a Local Supernova
To appear in Space Exploration Research, Editors: J. H. Denis, P. D. Aldridge Chapter 7 (Nova Science Publishers, Inc.) ISBN: 978-1-60692-264-4 Fingerprints of a Local Supernova Oliver Manuela and Hilton Ratcliffeb a Nuclear Chemistry, University of Missouri, Rolla, MO 65401 USA b Climate and Solar Science Institute, Cape Girardeau, MO 63701 USA ABSTRACT. The results of precise analysis of elements and isotopes in meteorites, comets, the Earth, the Moon, Mars, Jupiter, the solar wind, solar flares, and the solar photosphere since 1960 reveal the fingerprints of a local supernova (SN)—undiluted by interstellar material. Heterogeneous SN debris formed the planets. The Sun formed on the neutron (n) rich SN core. The ground-state masses of nuclei reveal repulsive n-n interactions that can trigger axial n-emission and a series of nuclear reactions that generate solar luminosity, the solar wind, and the measured flux of solar neutrinos. The location of the Sun's high-density core shifts relative to the solar surface as gravitational forces exerted by the major planets cause the Sun to experience abrupt acceleration and deceleration, like a yoyo on a string, in its orbit about the ever-changing centre-of-mass of the solar system. Solar cycles (surface magnetic activity, solar eruptions, and sunspots) and major climate changes arise from changes in the depth of the energetic SN core remnant in the interior of the Sun. Keywords: supernovae; stellar systems; supernova debris; supernova remnants; origin, formation, abundances of elements; planetary -

From the Natural Transmutations of Uranium to Its Artificial Fission
O T T O H AH N From the natural transmutations of uranium to its artificial fission Nobel Lecture, December 13, 1946 The year 1946 marked a jubilee in the history of the chemical element, ura- nium. Fifty years earlier, in the spring of 1896, Henri Becquerel had discov- ered the remarkable radiation phenomena of this element, which were at that time grouped together under the name of radioactivity. For more than 100 years, uranium, discovered by W. H. Klaproth in 1789, had had a quiet existence as a somewhat rare but not particularly interesting element. After its inclusion in the Periodic System by D. Mendeleev and Lothar Meyer, it was distinguished from all the other elements in one partic- ular respect: it occupied the highest place in the table of the elements. As yet, however, that did not have any particular significance. We know today that it is just this position of uranium at the highest place of the then known chemical elements which gives it the important properties by which it is distinguished from all other elements. The echo of Becquerel’s fundamental observations on the radioactivity of uranium in scientific circles was at first fairly weak. Two years later, how- ever, they acquired an exceptional importance when the Curies succeeded in separating from uranium minerals two active substances, polonium and ra- dium, of which the latter appeared to be several million times stronger than the same weight of uranium. It was only a few years before the first surprising property of this "ra- diating" substance was explained. -

Modern Physics
REVIEWS OF MODERN PHYSICS VoLUME 29, NuMBER 4 OcroBER, 1957 Synthesis of the Elements in Stars* E. MARGARET BURBIDGE, G. R. BURBIDGE, WILLIAM: A. FOWLER, AND F. HOYLE Kellogg Radiation1Laboratory, California Institute of Technology, and M aunt Wilson and Palomar Observatories, Carnegie Institution of Washington, California Institute of Technology, Pasadena, California "It is the stars, The stars above us, govern our conditions"; (King Lear, Act IV, Scene 3) but perhaps "The fault, dear Brutus, is not in our stars, But in ourselves," (Julius Caesar, Act I, Scene 2) TABLE OF CONTENTS Page I. Introduction ...............................................................· ............... 548 A. Element Abundances and Nuclear Structure. 548 B. Four Theories of the Origin of the Elements ............................................... 550 C. General Features of Stellar Synthesis ..................................................... 550 II. Physical Processes Involved in Stellar Synthesis, Their Place of Occurrence, and the Time-Scales Associated with Them ...............· ..................... · ................................. 551 A. Modes of Element Synthesis ............................................................. 551 B. Method of Assignment of Isotopes among Processes (i) to (viii) .............................. 553 C. Abundances and Synthesis Assignments Given in the Appendix. 555 D. Time-Scales for Different Modes of Synthesis .............................................. 556 III. Hydrogen Burning, Helium Burning, the a Process, -

Discovery of the Barium Isotopes
Preprint submitted to ATOMIC DATA AND NUCLEAR DATA TABLES May 12, 2009 Discovery of the Barium Isotopes A. SHORE, A. FRITSCH, J.Q. GINEPRO, M. HEIM, A. SCHUH, and M. THOENNESSEN ∗, National Superconducting Cyclotron Laboratory and Department of Physics and Astronomy, Michigan State University, East Lansing, MI 48824, USA Thirty-eight barium isotopes have so far been observed; the discovery of these isotopes is discussed. For each isotope a brief summary of the first refereed publication, including the production and identification method, is presented. ∗ Corresponding author. Email address: [email protected] (M. Thoennessen). CONTENTS 1 Introduction . 2 2 Discovery of 114−151Ba ............................................................. 2 3 Summary......................................................................... 12 EXPLANATION OF TABLE . 16 TABLE I. Discovery of Barium Isotopes . 17 REFERENCES FOR TABLE . 19 1. INTRODUCTION The tenth paper in the series of the discovery of isotopes, the discovery of the barium isotopes is discussed. Previously, the discoveries of cerium [1], arsenic [2], gold [3], tungsten [4], krypton [5], ein- steinium [6], iron [7], vanadium [8], and silver [9] isotopes were discussed. The purpose of this series is to document and summarize the discovery of the isotopes. Guidelines for assigning credit for discov- ery are (1) clear identification, either through decay-curves and relationships to other known isotopes, particle or g-ray spectra, or unique mass and Z-identification, and (2) publication of the discovery in a refereed journal. The authors and year of the first publication, the laboratory where the isotopes were produced as well as the production and identification methods are discussed. When appropriate, refer- ences to conference proceedings, internal reports, and theses are included. -
Meteoritic Barium and Anomalies Cerium Versus in Meteoritic The
Geochemical Journal, Vol. 13, pp. 137 to 140, 1979 137 NOTE Meteoritic barium and cerium versus the general isotopic anomalies in meteoritic xenon P. K. KURODA Department of Chemistry, University of Arkansas, Fayetteville, Arkansas 72702 U.S.A. (Received March 10, 1979) The difference in the isotopic compositions of barium from some meteorites and from the terrestrial barium is explained as due to the alteration of the isotopic ratios by a combined effect of mass-dependent fractionation, neutron-capture and cosmic-ray irradiation processes, which took place prior to and during the period of formation of the solar system. The abundances of 134Ba and ' 35 Ba in the meteorite Bruderheim seem to be slightly enhanced due to the decays of 2.06-year ' 34Cs and 2.3 X 106-year 13 5Ba A small difference in the isotopic compositions of cerium in the Bruderheim meteorite and the terrestiral sample can also be attributed to neutron-capture processes, which occurred during an early irradiation period. It is shown that the barium and xenon isotopic anomalies observed in meteorites are closely related to each other. INTRODUCTION Sixteen years later, MCCULLOCHand WASSER BURG (1978) found isotopic anomalies for Soon after the discovery of the so-called barium and neodymium in two inclusions from special and general anomalies in meteoritic the Allende meteorite. They reported that xenon was made in 1960, KRUMENACHERet al. sample EKI-4-1 showed large positive excesses (1962) have reported that the barium from the in the unshielded isotopes 135Ba and 137Ba of Richardton chondrite was of normal isotopic 13.4 and 12.3 parts in 104, respectively, and con composition. -

Toxicological Profile for Cesium
TOXICOLOGICAL PROFILE FOR CESIUM U.S. DEPARTMENT OF HEALTH AND HUMAN SERVICES Public Health Service Agency for Toxic Substances and Disease Registry April 2004 CESIUM ii DISCLAIMER The use of company or product name(s) is for identification only and does not imply endorsement by the Agency for Toxic Substances and Disease Registry. CESIUM iii UPDATE STATEMENT A Toxicological Profile for Cesium, Draft for Public Comment was released in July 2001. This edition supersedes any previously released draft or final profile. Toxicological profiles are revised and republished as necessary. For information regarding the update status of previously released profiles, contact ATSDR at: Agency for Toxic Substances and Disease Registry Division of Toxicology/Toxicology Information Branch 1600 Clifton Road NE, Mailstop E-32 Atlanta, Georgia 30333 vi Background Information The toxicological profiles are developed by ATSDR pursuant to Section 104(i) (3) and (5) of the Comprehensive Environmental Response, Compensation, and Liability Act of 1980 (CERCLA or Superfund) for hazardous substances found at Department of Energy (DOE) waste sites. CERCLA directs ATSDR to prepare toxicological profiles for hazardous substances most commonly found at facilities on the CERCLA National Priorities List (NPL) and that pose the most significant potential threat to human health, as determined by ATSDR and the EPA. ATSDR and DOE entered into a Memorandum of Understanding on November 4, 1992 which provided that ATSDR would prepare toxicological profiles for hazardous substances based upon ATSDR=s or DOE=s identification of need. The current ATSDR priority list of hazardous substances at DOE NPL sites was announced in the Federal Register on July 24, 1996 (61 FR 38451). -
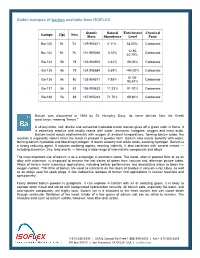
Stable Isotopes of Barium Available from ISOFLEX
Stable isotopes of barium available from ISOFLEX Atomic Natural Enrichment Chemical Isotope Z(p) N(n) Mass Abundance Level Form Ba-130 56 74 129.906311 0.11% 33.00% Carbonate 12.80- Ba-132 56 76 131.905056 0.10% Carbonate 40.70% Ba-134 56 78 133.904504 2.42% 88.00% Carbonate Ba-135 56 79 134.905684 6.59% >94.00% Carbonate 61.00- Ba-136 56 80 135.904571 7.85% Carbonate 95.40% Ba-137 56 81 136.905822 11.23% 91.70% Carbonate Ba-138 56 82 137.905242 71.70% 99.80% Carbonate Barium was discovered in 1808 by Sir Humphry Davy. Its name derives from the Greek word barys, meaning "heavy." A silvery-white, soft, ductile and somewhat malleable metal, barium gives off a green color in flame. It is extremely reactive and readily reacts with water, ammonia, halogens, oxygen and most acids. Barium metal reacts exothermically with oxygen at ambient temperatures, forming barium oxide; the reaction is especially violent when the metal is present in powder form. Barium also reacts violently with water, forming barium hydroxide and liberating hydrogen. It reacts violently with dilute acids, evolving hydrogen. Barium is a strong reducing agent. It reduces oxidizing agents, reacting violently. It also combines with several metals — including aluminum, zinc, lead and tin — forming a wide range of intermetallic compounds and alloys. The most important use of barium is as a scavenger in electronic tubes. The metal, often in powder form or as an alloy with aluminum, is employed to remove the last traces of gases from vacuum and television picture tubes. -
![Arxiv:1012.2027V1 [Nucl-Ex] 9 Dec 2010](https://docslib.b-cdn.net/cover/0671/arxiv-1012-2027v1-nucl-ex-9-dec-2010-6890671.webp)
Arxiv:1012.2027V1 [Nucl-Ex] 9 Dec 2010
Discovery of Zinc, Selenium, Bromine, and Neodymium Isotopes J. L. Gross, J. Claes, J. Kathawa, M. Thoennessen∗ National Superconducting Cyclotron Laboratory and Department of Physics and Astronomy, Michigan State University, East Lansing, MI 48824, USA Abstract Currently, thirty-two zinc, thirty-two selenium, twenty-nine bromine and thirty-one neodymium isotopes have been observed and the discovery of these isotopes is discussed here. For each isotope a brief synopsis of the first refereed publication, including the production and identification method, is presented. arXiv:1012.2027v1 [nucl-ex] 9 Dec 2010 ∗Corresponding author. Email address: [email protected] (M. Thoennessen) Preprint submitted to Atomic Data and Nuclear Data Tables November 14, 2018 Contents 1. Introduction . 4 2. Discovery of 54−85Zn ................................................................................... 5 2.1. 54Zn ............................................................................................ 5 2.2. 55;56Zn .......................................................................................... 7 2.3. 57Zn ............................................................................................ 7 2.4. 58Zn ............................................................................................ 7 2.5. 59Zn ............................................................................................ 7 2.6. 60;61Zn .......................................................................................... 7 2.7. 62Zn ...........................................................................................