• Benzene Does Not Undergo Electrophilic Addition • It Undergoes
Total Page:16
File Type:pdf, Size:1020Kb

Load more
Recommended publications
-

Products from Reactions of Carbon Nucleophiles and Carbon
14C synthesis strategies, Chem 315/316 / Beauchamp 1 Products from reactions of carbon nucleophiles and carbon electrophiles used in the 14C Game and our course: Carbon and hydrogen nucleophiles Ph R Ph Al H N R Li R (MgBr) P Li AlH R Cu Li Na CN RCC Na Ph 4 Li Carbon 2 H C R organolithium organolithium cyanide acetylides 2 ylid Na BH4 diisobutylaluminium lithium diisopropy- electrophiles cuprates (LAH) o reagents reagents Wittig reagents hydride (DIBAH) amide (LDA), -78 C H C Br 3 not useful not useful 2 RX coupling nitrilesalkynes not useful alkyls alkyls not useful methyl RX reaction R Br not useful not useful 2 RX coupling nitriles alkynes not useful alkyls alkyls not useful primary RX reaction R 2 RX coupling nitriles alkyls not useful E2 not useful alkyls not useful not useful reaction R Br secondary RX O 1o ROH 1o ROH 1o ROH 1o ROH not useful 1o ROH o not useful not useful 1o ROH 1 ROH ethylene oxide nitriles alkynes O o o o o o o 2 ROH 2 ROH 2 ROH 2 ROH 2o ROH 2 ROH 2 ROH not useful not useful E2, make nitriles alkynes allylic alcohols propylene oxide O o 3 ROH 3o ROH o 3o ROH o not useful o 3o ROH not useful E2, make 3 ROH 3 ROH 3 ROH allylic alcohols nitriles alkynes isobutylene oxide O o o specific methanol methanol C 1 ROH 1 ROH not used cyanohydrin 1o ROH not useful not useful in our course alkenes H H alkynes methanal O specific o enolate o o cyanohydrin o 1 ROH o C 2 ROH 2 ROH not useful 2 ROH alkenes 1 ROH not useful chemistry R H alkynes simple aldehydes O cyanohydrin o unless specific o enolate C 3 ROH 3o ROH o 2o ROH -

Organic Chemistry
Wisebridge Learning Systems Organic Chemistry Reaction Mechanisms Pocket-Book WLS www.wisebridgelearning.com © 2006 J S Wetzel LEARNING STRATEGIES CONTENTS ● The key to building intuition is to develop the habit ALKANES of asking how each particular mechanism reflects Thermal Cracking - Pyrolysis . 1 general principles. Look for the concepts behind Combustion . 1 the chemistry to make organic chemistry more co- Free Radical Halogenation. 2 herent and rewarding. ALKENES Electrophilic Addition of HX to Alkenes . 3 ● Acid Catalyzed Hydration of Alkenes . 4 Exothermic reactions tend to follow pathways Electrophilic Addition of Halogens to Alkenes . 5 where like charges can separate or where un- Halohydrin Formation . 6 like charges can come together. When reading Free Radical Addition of HX to Alkenes . 7 organic chemistry mechanisms, keep the elec- Catalytic Hydrogenation of Alkenes. 8 tronegativities of the elements and their valence Oxidation of Alkenes to Vicinal Diols. 9 electron configurations always in your mind. Try Oxidative Cleavage of Alkenes . 10 to nterpret electron movement in terms of energy Ozonolysis of Alkenes . 10 Allylic Halogenation . 11 to make the reactions easier to understand and Oxymercuration-Demercuration . 13 remember. Hydroboration of Alkenes . 14 ALKYNES ● For MCAT preparation, pay special attention to Electrophilic Addition of HX to Alkynes . 15 Hydration of Alkynes. 15 reactions where the product hinges on regio- Free Radical Addition of HX to Alkynes . 16 and stereo-selectivity and reactions involving Electrophilic Halogenation of Alkynes. 16 resonant intermediates, which are special favor- Hydroboration of Alkynes . 17 ites of the test-writers. Catalytic Hydrogenation of Alkynes. 17 Reduction of Alkynes with Alkali Metal/Ammonia . 18 Formation and Use of Acetylide Anion Nucleophiles . -

Chapter 7, Haloalkanes, Properties and Substitution Reactions of Haloalkanes Table of Contents 1
Chapter 7, Haloalkanes, Properties and Substitution Reactions of Haloalkanes Table of Contents 1. Alkyl Halides (Haloalkane) 2. Nucleophilic Substitution Reactions (SNX, X=1 or 2) 3. Nucleophiles (Acid-Base Chemistry, pka) 4. Leaving Groups (Acid-Base Chemistry, pka) 5. Kinetics of a Nucleophilic Substitution Reaction: An SN2 Reaction 6. A Mechanism for the SN2 Reaction 7. The Stereochemistry of SN2 Reactions 8. A Mechanism for the SN1 Reaction 9. Carbocations, SN1, E1 10. Stereochemistry of SN1 Reactions 11. Factors Affecting the Rates of SN1 and SN2 Reactions 12. --Eliminations, E1 and E2 13. E2 and E1 mechanisms and product predictions In this chapter we will consider: What groups can be replaced (i.e., substituted) or eliminated The various mechanisms by which such processes occur The conditions that can promote such reactions Alkyl Halides (Haloalkane) An alkyl halide has a halogen atom bonded to an sp3-hybridized (tetrahedral) carbon atom The carbon–chlorine and carbon– bromine bonds are polarized because the halogen is more electronegative than carbon The carbon-iodine bond do not have a per- manent dipole, the bond is easily polarizable Iodine is a good leaving group due to its polarizability, i.e. its ability to stabilize a charge due to its large atomic size Generally, a carbon-halogen bond is polar with a partial positive () charge on the carbon and partial negative () charge on the halogen C X X = Cl, Br, I Different Types of Organic Halides Alkyl halides (haloalkanes) sp3-hybridized Attached to Attached to Attached -

Chemical Kinetics HW1 (Kahn, 2010)
Chemical Kinetics HW1 (Kahn, 2010) Question 1. (6 pts) A reaction with stoichiometry A = P + 2Q was studied by monitoring the concentration of the reactant A as a function of time for eighteen minutes. The concentration determination method had a maximum error of 6 M. The following concentration profile was observed: Time (min) Conc (mM) 1 0.9850 2 0.8571 3 0.7482 4 0.6549 5 0.5885 6 0.5183 7 0.4667 8 0.4281 9 0.3864 10 0.3557 11 0.3259 12 0.3037 13 0.2706 14 0.2486 15 0.2355 16 0.2188 17 0.2111 18 0.1930 Determine the reaction order and calculate the rate constant for decomposition of A. What can be said about the mechanism or molecularity of this reaction? Question 2. (4 pts) Solve problem 2 on pg 31 in your textbook (House) using both linear and non-linear regression. Provide standard errors for the rate constant and half-life based on linear and non-linear fits. Below is the data set for your convenience: dataA = {{0, 0.5}, {10, 0.443}, {20,0.395}, {30,0.348}, {40,0.310}, {50,0.274}, {60,0.24}, {70,0.212}, {80,0.190}, {90,0.171}, {100,0.164}} Question 3. (3 pts) Solve problem 3 on pg 32 in your textbook (House). Question 4. (7 pts) The authors of the paper “Microsecond Folding of the Cold Shock Protein Measured by a Pressure-Jump Technique” suggest that the activated state of folding of CspB follows Hammond- type behavior. -

Reactions of Benzene & Its Derivatives
Organic Lecture Series ReactionsReactions ofof BenzeneBenzene && ItsIts DerivativesDerivatives Chapter 22 1 Organic Lecture Series Reactions of Benzene The most characteristic reaction of aromatic compounds is substitution at a ring carbon: Halogenation: FeCl3 H + Cl2 Cl + HCl Chlorobenzene Nitration: H2 SO4 HNO+ HNO3 2 + H2 O Nitrobenzene 2 Organic Lecture Series Reactions of Benzene Sulfonation: H 2 SO4 HSO+ SO3 3 H Benzenesulfonic acid Alkylation: AlX3 H + RX R + HX An alkylbenzene Acylation: O O AlX H + RCX 3 CR + HX An acylbenzene 3 Organic Lecture Series Carbon-Carbon Bond Formations: R RCl AlCl3 Arenes Alkylbenzenes 4 Organic Lecture Series Electrophilic Aromatic Substitution • Electrophilic aromatic substitution: a reaction in which a hydrogen atom of an aromatic ring is replaced by an electrophile H E + + + E + H • In this section: – several common types of electrophiles – how each is generated – the mechanism by which each replaces hydrogen 5 Organic Lecture Series EAS: General Mechanism • A general mechanism slow, rate + determining H Step 1: H + E+ E El e ctro - Resonance-stabilized phile cation intermediate + H fast Step 2: E + H+ E • Key question: What is the electrophile and how is it generated? 6 Organic Lecture Series + + 7 Organic Lecture Series Chlorination Step 1: formation of a chloronium ion Cl Cl + + - - Cl Cl+ Fe Cl Cl Cl Fe Cl Cl Fe Cl4 Cl Cl Chlorine Ferric chloride A molecular complex An ion pair (a Lewis (a Lewis with a positive charge containing a base) acid) on ch lorine ch loronium ion Step 2: attack of -

2 Reactions Observed with Alkanes Do Not Occur with Aromatic Compounds 2 (SN2 Reactions Never Occur on Sp Hybridized Carbons!)
Reactions of Aromatic Compounds Aromatic compounds are stabilized by this “aromatic stabilization” energy Due to this stabilization, normal SN2 reactions observed with alkanes do not occur with aromatic compounds 2 (SN2 reactions never occur on sp hybridized carbons!) In addition, the double bonds of the aromatic group do not behave similar to alkene reactions Aromatic Substitution While aromatic compounds do not react through addition reactions seen earlier Br Br Br2 Br2 FeBr3 Br With an appropriate catalyst, benzene will react with bromine The product is a substitution, not an addition (the bromine has substituted for a hydrogen) The product is still aromatic Electrophilic Aromatic Substitution Aromatic compounds react through a unique substitution type reaction Initially an electrophile reacts with the aromatic compound to generate an arenium ion (also called sigma complex) The arenium ion has lost aromatic stabilization (one of the carbons of the ring no longer has a conjugated p orbital) Electrophilic Aromatic Substitution In a second step, the arenium ion loses a proton to regenerate the aromatic stabilization The product is thus a substitution (the electrophile has substituted for a hydrogen) and is called an Electrophilic Aromatic Substitution Energy Profile Transition states Transition states Intermediate Potential E energy H Starting material Products E Reaction Coordinate The rate-limiting step is therefore the formation of the arenium ion The properties of this arenium ion therefore control electrophilic aromatic substitutions (just like any reaction consider the stability of the intermediate formed in the rate limiting step) 1) The rate will be faster for anything that stabilizes the arenium ion 2) The regiochemistry will be controlled by the stability of the arenium ion The properties of the arenium ion will predict the outcome of electrophilic aromatic substitution chemistry Bromination To brominate an aromatic ring need to generate an electrophilic source of bromine In practice typically add a Lewis acid (e.g. -

Chapter 20 the Carbonyl Can Be an Electrophile, It Can Be Attacked. It Can Occur Under Basic Or Acidic Conditions
Chapter 20 The carbonyl can be an electrophile, it can be attacked. It can occur under basic or acidic conditions. Basic Conditions: H O 1. Nu- O 2. H2O workup Nu Nu: HOH O : Nu Acidic Conditions: H+ H O : 1. H+, Nu: O Nu H + O OH Nu Nu: Hydride reductions. Basic conditions, H- is the nucleophile Ketones and aldehydes react w/ LiAlH4. H O 1. LiAlH4 O 2. H2O workup - H H: HOH O : H Esters and acid chlorides react w/ LiAlH4. H O 1. LiAlH4 O 2. H O workup OMe 2 H - H H: HOH O : O O : OMe H H H - H H: There are many sources of hydride with various reactivities Hydride Aldehyde Ketone Ester Amide Carboxyllic Acid Donors acid/ acid salt halide LiAlH4 Alcohol Alcohol Alcohol Amine Alcohol Alcohol LiAlH(OtBu)3 Alcohol Alcohol Alcohol - (too slow) N.R. Aldehyde NaBH4 Alcohol Alcohol N.R. N.R. N.R. ? NaBH3CN Alcohol N.R. N.R. N.R. N.R. ? DIBAL-H Alcohol Alcohol Aldehyde * Aldehyde* Alcohol ? * Stoichiometry must be carefully controlled. H2, Pd/C reacts preferentially with carbon-carbon double bonds. Carbon Nucleophiles: - Both of the following create reagents that act like CH3 . EtBr + 2Li →EtLi + LiBr EtBr + Mg → EtMgBr Alkyne nucleophiles. - + CH3C≡CH + NaNH2→ CH3C≡C: Na + NH3 - + CH3C≡CH + CH3Li → CH3C≡C: Li + CH4↑ CH3C≡CH + CH3MgBr → CH3C≡CMgBr + CH4↑ Ketones and aldehydes react w/ these carbon nucleophiles. H O 1. H3C CCMgBr O 2. H2O workup - C CH3CC: C CH HOH 3 O : Esters and acid chlorides react w/ these carbon nucleophiles. -

Using Carbon Dioxide As a Building Block in Organic Synthesis
REVIEW Received 10 Jul 2014 | Accepted 21 Nov 2014 | Published 20 Jan 2015 DOI: 10.1038/ncomms6933 Using carbon dioxide as a building block in organic synthesis Qiang Liu1, Lipeng Wu1, Ralf Jackstell1 & Matthias Beller1 Carbon dioxide exits in the atmosphere and is produced by the combustion of fossil fuels, the fermentation of sugars and the respiration of all living organisms. An active goal in organic synthesis is to take this carbon—trapped in a waste product—and re-use it to build useful chemicals. Recent advances in organometallic chemistry and catalysis provide effective means for the chemical transformation of CO2 and its incorporation into synthetic organic molecules under mild conditions. Such a use of carbon dioxide as a renewable one-carbon (C1) building block in organic synthesis could contribute to a more sustainable use of resources. more sensible resource management is the prerequisite for the sustainable development of future generations. However, when dealing with the feedstock of the chemical Aindustry, the level of sustainability is still far from satisfactory. Until now, the vast majority of carbon resources are based on crude oil, natural gas and coal. In addition to biomass, CO2 offers the possibility to create a renewable carbon economy. Since pre-industrial times, the amount of CO2 has steadily increased and nowadays CO2 is a component of greenhouse gases, which are primarily responsible for the rise in atmospheric temperature and probably abnormal changes in the global climate. This increase in CO2 concentration is largely due to the combustion of fossil fuels, which are required to meet the world’s energy demand1. -

Reactions of Aromatic Compounds Just Like an Alkene, Benzene Has Clouds of Electrons Above and Below Its Sigma Bond Framework
Reactions of Aromatic Compounds Just like an alkene, benzene has clouds of electrons above and below its sigma bond framework. Although the electrons are in a stable aromatic system, they are still available for reaction with strong electrophiles. This generates a carbocation which is resonance stabilized (but not aromatic). This cation is called a sigma complex because the electrophile is joined to the benzene ring through a new sigma bond. The sigma complex (also called an arenium ion) is not aromatic since it contains an sp3 carbon (which disrupts the required loop of p orbitals). Ch17 Reactions of Aromatic Compounds (landscape).docx Page1 The loss of aromaticity required to form the sigma complex explains the highly endothermic nature of the first step. (That is why we require strong electrophiles for reaction). The sigma complex wishes to regain its aromaticity, and it may do so by either a reversal of the first step (i.e. regenerate the starting material) or by loss of the proton on the sp3 carbon (leading to a substitution product). When a reaction proceeds this way, it is electrophilic aromatic substitution. There are a wide variety of electrophiles that can be introduced into a benzene ring in this way, and so electrophilic aromatic substitution is a very important method for the synthesis of substituted aromatic compounds. Ch17 Reactions of Aromatic Compounds (landscape).docx Page2 Bromination of Benzene Bromination follows the same general mechanism for the electrophilic aromatic substitution (EAS). Bromine itself is not electrophilic enough to react with benzene. But the addition of a strong Lewis acid (electron pair acceptor), such as FeBr3, catalyses the reaction, and leads to the substitution product. -

Reaction Mechanism Lecture 3 : Nucleophilic And
Module 10 : Reaction mechanism Lecture 3 : Nucleophilic and Electrophilic Addition and Abstraction Objectives In this lecture you will learn the following Ligand activation by metal that leads to a direct external attack at the ligand. Nucleophilic addition and nucleophilic abstraction reactions. Electrophilic addition and electrophilic abstraction reactions. The nucleophilic and electrophilic substitution and abstraction reactions can be viewed as ways of activation of substrates to allow an external reagent to directly attack the metal activated ligand without requiring prior binding of the external reagent to the metal. The attacking reagent may be a nucleophile or an electrophile. The nucleophilic attack of the external reagent is favored if the LnM fragment is a poor π−base and a good σ−acid i.e., when the complex is cationic and/or when the other metal bound ligands are electron withdrawing such that the ligand getting activated gets depleted of electron density and can undergo an external attack by a nucleophile Nu−, like LiMe or OH−. The attack of the nucleophiles may result in the formation of a bond between the nucleophiles and the activated unsaturated substrate, in which case it is called nucleophilic addition, or may result in an abstraction of a part or the whole of the activated ligand, in which case it is called the nucleophilic abstraction. The nucleophilic addition and the abstraction reactions are discussed below. Nucleophilic addition An example of a nucleophilic addition reaction is shown below. Carbon monoxide (CO) as a ligand can undergo nucleophilic attack when bound to a metal center of poor π−basicity, as the carbon center of the CO ligand is electron deficient owing to the ligand to metal σ−donation not being fully compensated by the metal to ligand π−back donation. -
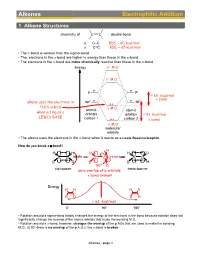
Alkenes Electrophilic Addition
Alkenes Electrophilic Addition 1 Alkene Structures chemistry of C C double bond σ C–C BDE ~ 80 kcal/mol π C=C BDE ~ 65 kcal/mol • The p-bond is weaker than the sigma-bond • The, electrons in the p-bond are higher in energy than those in the s-bond • The electrons in the p-bond are more chemically reactive than those in the s-bond Energy σ∗ M.O. π∗ M.O. p p ~ 65 kcal/mol 2 π bond alkene uses the electrons in sp2 sp THIS orbital π M.O. atomic atomic when acting as a orbitals orbitals ~ 81 kcal/mol LEWIS BASE carbon 1 carbon 2 σ bond σ M.O. molecular orbitals • The alkene uses the electrons in the p-bond when it reacts as a Lewis Base/nucleophile How do you break a p-bond? H H H H H D C C rotate C C rotate C C D D D D H 90° D cis-isomer trans-isomer zero overlap of p orbitals: π bond broken! Energy H H H D D D D H ~ 63 kcal/mol 0° 90° 180° • Rotation around a sigma-bond hardly changes the energy of the electrons in the bond because rotation does not significantly change the overlap of the atomic orbitals that make the bonding M.O. • Rotation around a p-bond, however, changes the overlap of the p AOs that are used to make the bonding M.O., at 90° there is no overlap of the p A.O.s, the p-bond is broken Alkenes : page 1 Distinguishing isomers trans- cis- • By now we are very familiar with cis- and trans-stereoisomers (diastereomers) • But what about, the following two structures, they can NOT be assigned as cis- or trans-, yet they are definitely stereoisomers (diastereomers), the directions in which their atoms point in space are different cis- / trans- Br Br We Need a different system to distinguish stereoisomers for C=C double bonds: Use Z/E notation. -

Reactions of Alkenes and Alkynes
05 Reactions of Alkenes and Alkynes Polyethylene is the most widely used plastic, making up items such as packing foam, plastic bottles, and plastic utensils (top: © Jon Larson/iStockphoto; middle: GNL Media/Digital Vision/Getty Images, Inc.; bottom: © Lakhesis/iStockphoto). Inset: A model of ethylene. KEY QUESTIONS 5.1 What Are the Characteristic Reactions of Alkenes? 5.8 How Can Alkynes Be Reduced to Alkenes and 5.2 What Is a Reaction Mechanism? Alkanes? 5.3 What Are the Mechanisms of Electrophilic Additions HOW TO to Alkenes? 5.1 How to Draw Mechanisms 5.4 What Are Carbocation Rearrangements? 5.5 What Is Hydroboration–Oxidation of an Alkene? CHEMICAL CONNECTIONS 5.6 How Can an Alkene Be Reduced to an Alkane? 5A Catalytic Cracking and the Importance of Alkenes 5.7 How Can an Acetylide Anion Be Used to Create a New Carbon–Carbon Bond? IN THIS CHAPTER, we begin our systematic study of organic reactions and their mecha- nisms. Reaction mechanisms are step-by-step descriptions of how reactions proceed and are one of the most important unifying concepts in organic chemistry. We use the reactions of alkenes as the vehicle to introduce this concept. 129 130 CHAPTER 5 Reactions of Alkenes and Alkynes 5.1 What Are the Characteristic Reactions of Alkenes? The most characteristic reaction of alkenes is addition to the carbon–carbon double bond in such a way that the pi bond is broken and, in its place, sigma bonds are formed to two new atoms or groups of atoms. Several examples of reactions at the carbon–carbon double bond are shown in Table 5.1, along with the descriptive name(s) associated with each.