Differentiating Drug‐Induced Multichannel Block on The
Total Page:16
File Type:pdf, Size:1020Kb

Load more
Recommended publications
-
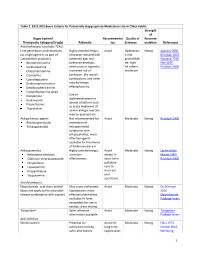
Table 2. 2012 AGS Beers Criteria for Potentially
Table 2. 2012 AGS Beers Criteria for Potentially Inappropriate Medication Use in Older Adults Strength of Organ System/ Recommendat Quality of Recomm Therapeutic Category/Drug(s) Rationale ion Evidence endation References Anticholinergics (excludes TCAs) First-generation antihistamines Highly anticholinergic; Avoid Hydroxyzin Strong Agostini 2001 (as single agent or as part of clearance reduced with e and Boustani 2007 combination products) advanced age, and promethazi Guaiana 2010 Brompheniramine tolerance develops ne: high; Han 2001 Carbinoxamine when used as hypnotic; All others: Rudolph 2008 Chlorpheniramine increased risk of moderate Clemastine confusion, dry mouth, Cyproheptadine constipation, and other Dexbrompheniramine anticholinergic Dexchlorpheniramine effects/toxicity. Diphenhydramine (oral) Doxylamine Use of diphenhydramine in Hydroxyzine special situations such Promethazine as acute treatment of Triprolidine severe allergic reaction may be appropriate. Antiparkinson agents Not recommended for Avoid Moderate Strong Rudolph 2008 Benztropine (oral) prevention of Trihexyphenidyl extrapyramidal symptoms with antipsychotics; more effective agents available for treatment of Parkinson disease. Antispasmodics Highly anticholinergic, Avoid Moderate Strong Lechevallier- Belladonna alkaloids uncertain except in Michel 2005 Clidinium-chlordiazepoxide effectiveness. short-term Rudolph 2008 Dicyclomine palliative Hyoscyamine care to Propantheline decrease Scopolamine oral secretions. Antithrombotics Dipyridamole, oral short-acting* May -

Dofetilide (Tikosyn): a New Drug to Control Atrial Fibrillation
CURRENT DRUG THERAPY WALID I. SALIBA, MD Section of Cardiac Electrophysiology and Pacing, CREDIT Department of Cardiology, Cleveland Clinic Dofetilide (Tikosyn): A new drug to control atrial fibrillation ABSTRACT OFETILIDE (Tikosyn), a new antiarrhyth- mic drug, can convert atrial fibrillation Dofetilide, a new class III antiarrhythmic agent, selectively and atrial flutter to sinus rhythm in approxi- blocks a specific cardiac potassium channel, lKr, increasing mately 30% of cases and maintain sinus the effective refractory period of the myocyte and thereby rhythm after electrical or pharmacologic con- terminating reentrant arrhythmias. Given orally, it appears version for up to 1 year in 60% to 70% of to effectively convert atrial fibrillation and atrial flutter to cases, without increasing the risk of sudden sinus rhythm and maintain sinus rhythm after conversion in death in patients at high risk. appropriately selected patients. This paper reviews the Such new drugs are needed, as many of the pharmacology of dofetilide, the evidence of its antiarrhythmic drugs in use up to now have effectiveness, and the appropriate precautions in using it. actually produced higher mortality rates in clinical trials than did placebo, or cause unac- ceptable side effects. KEY POINTS This article reviews the mechanism of action, safety, effectiveness, and clinical use of Dofetilide is generally well tolerated but like other class III dofetilide. drugs can cause torsades de pointes. The risk is dose- dependent and can be minimized by adjusting the dosage • PROBLEMS WITH PREVIOUS DRUGS according to creatinine clearance and QT interval, by excluding patients with known risk factors for long QT A variety of drugs have been used to terminate syndrome and torsades de pointes, and by starting or prevent atrial and ventricular arrhythmias, treatment in an inpatient monitored setting for the first 3 but their safety, efficacy, and tolerability in days. -

Safety and Efficacy of Ibutilide in Cardioversion of Atrial Flutter And
J Am Board Fam Med: first published as 10.3122/jabfm.2011.01.080096 on 5 January 2011. Downloaded from CLINICAL REVIEW Safety and Efficacy of Ibutilide in Cardioversion of Atrial Flutter and Fibrillation Madhuri Nair, MD, Lekha K. George, MD, and Santhosh K. G. Koshy, MD This article reviews the safety and efficacy of ibutilide for use in patients with atrial fibrillation and flut- ter. Ibutilide, a class III antiarrhythmic agent, is primarily used for conversion of atrial flutter and fi- brillation and is a good alternative to electrical cardioversion. Ibutilide has a conversion rate of up to 75% to 80% in recent-onset atrial fibrillation and flutter; the conversion rate is higher for atrial flutter than for atrial fibrillation. It is also safe in the conversion of chronic atrial fibrillation/flutter among patients receiving oral amiodarone therapy. Ibutilide pretreatment facilitates transthoracic defibrilla- tion and decreases the energy requirement of electrical cardioversion by both monophasic and biphasic shocks. Pretreatment with ibutilide before electrical defibrillation has a conversion rate of 100% com- pared with 72% with no pretreatment. Ibutilide is also safe and efficient in the treatment of atrial fibril- lation in patients who have had cardiac surgery, and in accessory pathway–mediated atrial fibrillation where the conversion rate of ibutilide is as high as 95%. There is up to a 4% risk of torsade de pointes and a 4.9% risk of monomorphic ventricular tachycardia. Hence, close monitoring in an intensive care unit setting is warranted during and at least for 4 hours after drug infusion. The anticoagulation strat- egy is the same as for any other mode of cardioversion.(J Am Board Fam Med 2011;24:86–92.) Keywords: Antiarrhythmics, Arrhythmia, Atrial Fibrillation, Cardiovascular Disorders, Cardioversion, Drug Ther- copyright. -

Cocaine Intoxication and Hypertension
THE EMCREG-INTERNATIONAL CONSENSUS PANEL RECOMMENDATIONS Cocaine Intoxication and Hypertension Judd E. Hollander, MD From the Department of Emergency Medicine, University of Pennsylvania, Philadelphia, PA. 0196-0644/$-see front matter Copyright © 2008 by the American College of Emergency Physicians. doi:10.1016/j.annemergmed.2007.11.008 [Ann Emerg Med. 2008;51:S18-S20.] with cocaine intoxication is analogous to that of the patient with hypertension: the treatment should be geared toward the Cocaine toxicity has been reported in virtually all organ patient’s presenting complaint. systems. Many of the adverse effects of cocaine are similar to When the medical history is clear and symptoms are mild, adverse events that can result from either acute hypertensive laboratory evaluation is usually unnecessary. In contrast, if the crisis or chronic effects of hypertension. Recognizing when the patient has severe toxicity, evaluation should be geared toward specific disease requires treatment separate from cocaine toxicity the presenting complaint. Laboratory evaluation may include a is paramount to the treatment of patients with cocaine CBC count; determination of electrolyte, glucose, blood urea intoxication. nitrogen, creatine kinase, and creatinine levels; arterial blood The initial physiologic effect of cocaine on the cardiovascular gas analysis; urinalysis; and cardiac marker determinations. system is a transient bradycardia as a result of stimulation of the Increased creatine kinase level occurs with rhabdomyolysis. vagal nuclei. Tachycardia typically ensues, predominantly from Cardiac markers are increased in myocardial infarction. Cardiac increased central sympathetic stimulation. Cocaine has a troponin I is preferred to identify acute myocardial13 infarction. cardiostimulatory effect through sensitization to epinephrine A chest radiograph should be obtained in patients with and norepinephrine. -

Abstract Materials and Methods Summary
A human ex-vivo contractility-based assay for the simultaneous prediction of drug-induced inotropic and pro-arrhythmia risk Najah Abi-Gerges, Ashley Alamillo, Phachareeya Ratchada, Guy Page, Yannick Miron, Nathalie Nguyen, Paul E Miller and Andre Ghetti AnaBios Corp., San Diego, CA 92109, USA Contact email: [email protected] Abstract Human ventricular trabeculae-based model predicts Human ventricular trabeculae-based model the torsadogenic potential of dofetilide and d,l-sotalol predicts safety of verapamil Cardiac safety remains the leading cause of drug development discontinuation and withdrawal of post marketing approvals. This has called into question the reliability of current preclinical safety- testing paradigms, which rely predominantly on animal models, and has led to demands for more predictive tools. To this aim, we sought to develop and validate a new ex-vivo human-based model that uses human ventricular trabeculae, paced under tension and Figure 3. Effects of verapamil on Fc parameters in human ex-vivo ventricular combined with continuous recordings of the exerted force of trabeculae. Verapamil decreased the force contraction (Fc). We were especially interested in establishing if of contraction with an IC50 value of 1.46µM, while it had no effect on TR70 and did not this strategy could provide a more predictive approach for induce any ACs or triggered contractions when tested up to 222-fold its fETPC. n=7 assessing both inotropic activity as well as pro-arrhythmia risk. trabeculae. Note that the negative inotropic Since the T-wave recorded in ECG in the clinics marks ventricular effect of verapamil is in line with its known 2+ repolarization and is therefore an electrical measure of the inhibition of Ca channel). -

Guideline for Preoperative Medication Management
Guideline: Preoperative Medication Management Guideline for Preoperative Medication Management Purpose of Guideline: To provide guidance to physicians, advanced practice providers (APPs), pharmacists, and nurses regarding medication management in the preoperative setting. Background: Appropriate perioperative medication management is essential to ensure positive surgical outcomes and prevent medication misadventures.1 Results from a prospective analysis of 1,025 patients admitted to a general surgical unit concluded that patients on at least one medication for a chronic disease are 2.7 times more likely to experience surgical complications compared with those not taking any medications. As the aging population requires more medication use and the availability of various nonprescription medications continues to increase, so does the risk of polypharmacy and the need for perioperative medication guidance.2 There are no well-designed trials to support evidence-based recommendations for perioperative medication management; however, general principles and best practice approaches are available. General considerations for perioperative medication management include a thorough medication history, understanding of the medication pharmacokinetics and potential for withdrawal symptoms, understanding the risks associated with the surgical procedure and the risks of medication discontinuation based on the intended indication. Clinical judgement must be exercised, especially if medication pharmacokinetics are not predictable or there are significant risks associated with inappropriate medication withdrawal (eg, tolerance) or continuation (eg, postsurgical infection).2 Clinical Assessment: Prior to instructing the patient on preoperative medication management, completion of a thorough medication history is recommended – including all information on prescription medications, over-the-counter medications, “as needed” medications, vitamins, supplements, and herbal medications. Allergies should also be verified and documented. -

Anaesthetic Implications of Calcium Channel Blockers
436 Anaesthetic implications of calcium channel Leonard C. Jenkins aA MD CM FRCPC blockers Peter J. Scoates a sc MD FRCPC CONTENTS The object of this review is to emphasize the anaesthetic implications of calcium channel block- Physiology - calcium/calcium channel blockers Uses of calcium channel blockers ers for the practising anaesthetist. These drugs have Traditional played an expanding role in therapeutics since their Angina pectoris introduction and thus anaesthetists can expect to see Arrhythmias increasing numbers of patients presenting for anaes- Hypertension thesia who are being treated with calcium channel Newer and investigational Cardiac blockers. Other reviews have emphasized the basic - Hypertrophic cardiomyopathy pharmacology of calcium channel blockers. 1-7 - Cold cardioplegia - Pulmonary hypertension Physiology - calcium/calcium channel blockers Actions on platelets Calcium plays an important role in many physio- Asthma Obstetrics logical processes, such as blood coagulation, en- - Premature labor zyme systems, muscle contraction, bone metabo- - Pre-eclampsia lism, synaptic transmission, and cell membrane Achalasia and oesophageal spasm excitability. Especially important is the role of Increased intraocular pressure therapy calcium in myocardial contractility and conduction Protective effect on kidney after radiocontrast Cerebral vasospasm as well as in vascular smooth muscle reactivity. 7 Induced hypotensive anaesthesia Thus, it can be anticipated that any drug interfering Drag interactions with calcium channel blockers with the action of calcium could have widespread With anaesthetic agents effects. Inhalation agents In order to understand the importance of calcium - Effect on haemodynamics - Effect on MAC in cellular excitation, it is necessary to review some Neuromuscular blockers membrane physiology. Cell membranes are pri- Effects on epinephrine-induced arrhythmias marily phospholipids arranged in a bilayer. -

Verapamil Inhibits TRESK (K2P18.1) Current in Trigeminal Ganglion Neurons Independently of the Blockade of Ca2+ Influx
International Journal of Molecular Sciences Article Verapamil Inhibits TRESK (K2P18.1) Current in Trigeminal Ganglion Neurons Independently of the Blockade of Ca2+ Influx Hyun Park 1, Eun-Jin Kim 2, Ji Hyeon Ryu 2,3, Dong Kun Lee 2,3, Seong-Geun Hong 2,3, Jaehee Han 2, Jongwoo Han 1,* and Dawon Kang 2,3,* ID 1 Department of Neurosurgery, College of Medicine and Institute of Health Sciences, Gyeongsang National University, Jinju 52727, South Korea; [email protected] 2 Department of Physiology and Institute of Health Sciences, College of Medicine, Gyeongsang National University, Jinju 52727, South Korea; [email protected] (E.-J.K.); [email protected] (J.H.R.); [email protected] (D.K.L.); [email protected] (S.-G.H.); [email protected] (J.H.) 3 Department of Convergence Medical Science, Gyeongsang National University, Jinju 52727, South Korea * Correspondence: [email protected] (J.H.); [email protected] (D.K.); Tel.: +82-55-772-8044 (D.K.) Received: 5 June 2018; Accepted: 2 July 2018; Published: 4 July 2018 Abstract: Tandem pore domain weak inward rectifier potassium channel (TWIK)-related spinal cord + + K (TRESK; K2P18.1) channel is the only member of the two-pore domain K (K2P) channel family 2+ 2+ that is activated by an increase in intracellular Ca concentration ([Ca ]i) and linked to migraines. This study was performed to identify the effect of verapamil, which is an L-type Ca2+ channel blocker and a prophylaxis for migraines, on the TRESK channel in trigeminal ganglion (TG) neurons, as well as in a heterologous system. -

Patent Application Publication ( 10 ) Pub . No . : US 2019 / 0192440 A1
US 20190192440A1 (19 ) United States (12 ) Patent Application Publication ( 10) Pub . No. : US 2019 /0192440 A1 LI (43 ) Pub . Date : Jun . 27 , 2019 ( 54 ) ORAL DRUG DOSAGE FORM COMPRISING Publication Classification DRUG IN THE FORM OF NANOPARTICLES (51 ) Int . CI. A61K 9 / 20 (2006 .01 ) ( 71 ) Applicant: Triastek , Inc. , Nanjing ( CN ) A61K 9 /00 ( 2006 . 01) A61K 31/ 192 ( 2006 .01 ) (72 ) Inventor : Xiaoling LI , Dublin , CA (US ) A61K 9 / 24 ( 2006 .01 ) ( 52 ) U . S . CI. ( 21 ) Appl. No. : 16 /289 ,499 CPC . .. .. A61K 9 /2031 (2013 . 01 ) ; A61K 9 /0065 ( 22 ) Filed : Feb . 28 , 2019 (2013 .01 ) ; A61K 9 / 209 ( 2013 .01 ) ; A61K 9 /2027 ( 2013 .01 ) ; A61K 31/ 192 ( 2013. 01 ) ; Related U . S . Application Data A61K 9 /2072 ( 2013 .01 ) (63 ) Continuation of application No. 16 /028 ,305 , filed on Jul. 5 , 2018 , now Pat . No . 10 , 258 ,575 , which is a (57 ) ABSTRACT continuation of application No . 15 / 173 ,596 , filed on The present disclosure provides a stable solid pharmaceuti Jun . 3 , 2016 . cal dosage form for oral administration . The dosage form (60 ) Provisional application No . 62 /313 ,092 , filed on Mar. includes a substrate that forms at least one compartment and 24 , 2016 , provisional application No . 62 / 296 , 087 , a drug content loaded into the compartment. The dosage filed on Feb . 17 , 2016 , provisional application No . form is so designed that the active pharmaceutical ingredient 62 / 170, 645 , filed on Jun . 3 , 2015 . of the drug content is released in a controlled manner. Patent Application Publication Jun . 27 , 2019 Sheet 1 of 20 US 2019 /0192440 A1 FIG . -

Verapamil 40Mg, 80Mg, 120Mg and 160Mg Tablets
6 Contents of the pack and other information What Verapamil tablets contain • The active substance is verapamil hydrochloride. Package leaflet: Information for the patient Each tablet contains either 40mg, 80mg, 120mg or 160mg of verapamil hydrochloride. • The other ingredients are croscarmellose Verapamil 40mg, 80mg, 120mg sodium, magnesium stearate, maize starch, and 160mg tablets propylene glycol, sunset yellow aluminium lake (E110), quinoline yellow aluminium lake Read all of this leaflet carefully before you • manage high blood pressure (hypertension), (E104), titanium dioxide (E171), microcrystalline start taking this medicine because it contains used alone or with other drugs for high blood cellulose (E460), hydroxypropylcellulose (E463), pressure. methylhydroxypropylcellulose (E464), purified talc important information for you. • manage and prevent angina. (E553). • Keep this leaflet. You may need to read it again. • treat and prevent certain types of abnormal heartbeats. What Verapamil tablets look like and contents of • If you have any further questions, ask your the pack doctor or pharmacist. 2 What you need to know before you Verapamil 40mg tablets are yellow, circular, biconvex, • This medicine has been prescribed for you film-coated tablets, impressed “C” on one face and only. Do not pass it on to others. It may harm take Verapamil tablets Do not take Verapamil tablets if you: the identifying letters “VR” on the reverse. Tablet them, even if their signs of illness are the • are allergic to verapamil hydrochloride or any of diameter: 6.18-6.82mm. same as yours. the other ingredients of this medicine (listed in Verapamil 80mg tablets are yellow, circular, biconvex • If you get any side effects, talk to your doctor film-coated tablets, impressed “C” on one face and section 6) or pharmacist. -

Calcium Channel Blockers
Calcium Channel Blockers Summary In general, calcium channel blockers (CCBs) are used most often for the management of hypertension and angina. There are 2 classes of CCBs: the dihydropyridines (DHPs), which have greater selectivity for vascular smooth muscle cells than for cardiac myocytes, and the non-DHPs, which have greater selectivity for cardiac myocytes and are used for cardiac arrhythmias. The DHPs cause peripheral edema, headaches, and postural hypotension most commonly, all of which are due to the peripheral vasodilatory effects of the drugs in this class of CCBs. The non-DHPs are negative inotropes and chronotropes; they can cause bradycardia and depress AV node conduction, increasing the risk of heart failure exacerbation, bradycardia, and AV block. Clevidipine is a DHP calcium channel blocker administered via continuous IV infusion and used for rapid blood pressure reductions. All CCBs are substrates of CYP3A4, but both diltiazem and verapamil are also inhibitors of 3A4 and have an increased risk of drug interactions. Verapamil also inhibits CYP2C9, CYP2C19, and CYP1A2. Pharmacology CCBs selectively inhibit the voltage-gated L-type calcium channels on cardiac myocytes, vascular smooth muscle cells, and cells within the sinoatrial (SA) and atrioventricular (AV) nodes, preventing influx of extracellular calcium. CCBs act by either deforming the channels, inhibiting ion-control gating mechanisms, and/or interfering with the release of calcium from the major cellular calcium store, the endoplasmic reticulum. Calcium influx via these channels serves for excitation-contraction coupling and electrical discharge in the heart and vasculature. A decrease in intracellular calcium will result in inhibition of the contractile process of the myocardial smooth muscle cells, resulting in dilation of the coronary and peripheral arterial vasculature. -

Randomized Study of Dofetilide, Quinidine, Ranolazine, and Verapamil
Clinical Trial nature publishing group CLINICAL TRIAL see COMMENTARY page 534 Differentiating Drug-Induced Multichannel Block on the Electrocardiogram: Randomized Study of Dofetilide, Quinidine, Ranolazine, and Verapamil L Johannesen1,2, J Vicente1,3, JW Mason4, C Sanabria4, K Waite-Labott4, M Hong5, P Guo5, J Lin5, JS Sørensen6, L Galeotti1, J Florian6, M Ugander1,2, N Stockbridge7 and DG Strauss1,2 Block of the hERG potassium channel and prolongation of the QT interval are predictors of drug-induced torsade de pointes. However, drugs that block the hERG potassium channel may also block other channels that mitigate torsade risk. We hypothesized that the electrocardiogram can differentiate the effects of multichannel drug block by separate analysis of early repolarization (global J–Tpeak) and late repolarization (global Tpeak–Tend). In this prospective randomized controlled clinical trial, 22 subjects received a pure hERG potassium channel blocker (dofetilide) and three drugs that block hERG and either calcium or late sodium currents (quinidine, ranolazine, and verapamil). The results show that hERG potassium channel block equally prolongs early and late repolarization, whereas additional inward current block (calcium or late sodium) preferentially shortens early repolarization. Characterization of multichannel drug effects on human cardiac repolarization is possible and may improve the utility of the electrocardiogram in the assessment of drug-related cardiac electrophysiology. Fourteen drugs have been removed from the market worldwide and/or sodium channels (inward currents). The most notable because they increase the risk for torsade de pointes,1 a ven- example is amiodarone, which causes substantial QT prolonga- tricular arrhythmia that can cause sudden cardiac death.