Download?Doi=10.1.1.822.6038&Rep=Rep1&Type=Pdf
Total Page:16
File Type:pdf, Size:1020Kb

Load more
Recommended publications
-

Mouse Mxra8 Knockout Project (CRISPR/Cas9)
https://www.alphaknockout.com Mouse Mxra8 Knockout Project (CRISPR/Cas9) Objective: To create a Mxra8 knockout Mouse model (C57BL/6J) by CRISPR/Cas-mediated genome engineering. Strategy summary: The Mxra8 gene (NCBI Reference Sequence: NM_024263 ; Ensembl: ENSMUSG00000029070 ) is located on Mouse chromosome 4. 10 exons are identified, with the ATG start codon in exon 1 and the TAA stop codon in exon 10 (Transcript: ENSMUST00000030947). Exon 1~10 will be selected as target site. Cas9 and gRNA will be co-injected into fertilized eggs for KO Mouse production. The pups will be genotyped by PCR followed by sequencing analysis. Note: Phenotypic analysis of mice homozygous for a gene trap allele indicates this mutation has no notable phenotype in any parameter tested in a high-throughput screen. Exon 1 starts from about 0.08% of the coding region. Exon 1~10 covers 100.0% of the coding region. The size of effective KO region: ~3486 bp. The KO region does not have any other known gene. Page 1 of 9 https://www.alphaknockout.com Overview of the Targeting Strategy Wildtype allele 5' gRNA region gRNA region 3' 1 2 3 4 5 6 7 8 9 10 Legends Exon of mouse Mxra8 Knockout region Page 2 of 9 https://www.alphaknockout.com Overview of the Dot Plot (up) Window size: 15 bp Forward Reverse Complement Sequence 12 Note: The 2000 bp section upstream of start codon is aligned with itself to determine if there are tandem repeats. No significant tandem repeat is found in the dot plot matrix. So this region is suitable for PCR screening or sequencing analysis. -

A Recombinant Virus and Reporter Mouse System to Study Chronic Chikungunya Virus Pathogenesis Alissa Roxanne Young Washington University in St
Washington University in St. Louis Washington University Open Scholarship Arts & Sciences Electronic Theses and Dissertations Arts & Sciences Winter 12-15-2018 A Recombinant Virus and Reporter Mouse System to Study Chronic Chikungunya Virus Pathogenesis Alissa Roxanne Young Washington University in St. Louis Follow this and additional works at: https://openscholarship.wustl.edu/art_sci_etds Part of the Allergy and Immunology Commons, Immunology and Infectious Disease Commons, Medical Immunology Commons, and the Virology Commons Recommended Citation Young, Alissa Roxanne, "A Recombinant Virus and Reporter Mouse System to Study Chronic Chikungunya Virus Pathogenesis" (2018). Arts & Sciences Electronic Theses and Dissertations. 1705. https://openscholarship.wustl.edu/art_sci_etds/1705 This Dissertation is brought to you for free and open access by the Arts & Sciences at Washington University Open Scholarship. It has been accepted for inclusion in Arts & Sciences Electronic Theses and Dissertations by an authorized administrator of Washington University Open Scholarship. For more information, please contact [email protected]. WASHINGTON UNIVERSITY IN ST. LOUIS Division of Biology and Biomedical Sciences Molecular Microbiology and Microbial PatHogenesis Dissertation Examination Committee: DeboraH J. Lenschow, Chair Adrianus C. M. Boon Michael S. Diamond Robyn S. Klein THaddeus S. StaPPenbeck David Wang A Recombinant Virus and RePorter Mouse System to Study CHronic CHikungunya Virus PatHogenesis by Alissa Roxanne Young A dissertation -

Human Induced Pluripotent Stem Cell–Derived Podocytes Mature Into Vascularized Glomeruli Upon Experimental Transplantation
BASIC RESEARCH www.jasn.org Human Induced Pluripotent Stem Cell–Derived Podocytes Mature into Vascularized Glomeruli upon Experimental Transplantation † Sazia Sharmin,* Atsuhiro Taguchi,* Yusuke Kaku,* Yasuhiro Yoshimura,* Tomoko Ohmori,* ‡ † ‡ Tetsushi Sakuma, Masashi Mukoyama, Takashi Yamamoto, Hidetake Kurihara,§ and | Ryuichi Nishinakamura* *Department of Kidney Development, Institute of Molecular Embryology and Genetics, and †Department of Nephrology, Faculty of Life Sciences, Kumamoto University, Kumamoto, Japan; ‡Department of Mathematical and Life Sciences, Graduate School of Science, Hiroshima University, Hiroshima, Japan; §Division of Anatomy, Juntendo University School of Medicine, Tokyo, Japan; and |Japan Science and Technology Agency, CREST, Kumamoto, Japan ABSTRACT Glomerular podocytes express proteins, such as nephrin, that constitute the slit diaphragm, thereby contributing to the filtration process in the kidney. Glomerular development has been analyzed mainly in mice, whereas analysis of human kidney development has been minimal because of limited access to embryonic kidneys. We previously reported the induction of three-dimensional primordial glomeruli from human induced pluripotent stem (iPS) cells. Here, using transcription activator–like effector nuclease-mediated homologous recombination, we generated human iPS cell lines that express green fluorescent protein (GFP) in the NPHS1 locus, which encodes nephrin, and we show that GFP expression facilitated accurate visualization of nephrin-positive podocyte formation in -

Complete Transcriptome Profiling of Normal and Age-Related Macular
www.nature.com/scientificreports OPEN Complete Transcriptome Profling of Normal and Age-Related Macular Degeneration Eye Tissues Reveals Received: 16 June 2017 Accepted: 30 January 2018 Dysregulation of Anti-Sense Published: xx xx xxxx Transcription Eun Ji Kim1, Gregory R. Grant1,2, Anita S. Bowman3,4, Naqi Haider3,4, Harini V. Gudiseva3 & Venkata Ramana Murthy Chavali3,4 Age-related macular degeneration (AMD) predominantly afects the retina and retinal pigment epithelium in the posterior eye. While there are numerous studies investigating the non-coding transcriptome of retina and RPE, few signifcant diferences between AMD and normal tissues have been reported. Strand specifc RNA sequencing of both peripheral retina (PR) and RPE-Choroid-Sclera (PRCS), in both AMD and matched normal controls were generated. The transcriptome analysis reveals a highly signifcant and consistent impact on anti-sense transcription as well as moderate changes in the regulation of non-coding (sense) RNA. Hundreds of genes that do not express anti-sense transcripts in normal PR and PRCS demonstrate signifcant anti-sense expression in AMD in all patient samples. Several pathways are highly enriched in the upregulated anti-sense transcripts—in particular the EIF2 signaling pathway. These results call for a deeper exploration into anti-sense and noncoding RNA regulation in AMD and their potential as therapeutic targets. Age-related macular degeneration (AMD) is the third largest cause of vision loss worldwide1. It is a progressive retinal disorder that involves loss of central vision, hypo- and hyper-pigmentation of the RPE, deposition of drusen in the Bruch’s membrane, and loss of photoreceptors, especially in the 8th or 9th decade2–4. -

The Tissue-Specific Transcriptomic Landscape of the Mid-Gestational Mouse Embryo Martin Werber1, Lars Wittler1, Bernd Timmermann2, Phillip Grote1,* and Bernhard G
© 2014. Published by The Company of Biologists Ltd | Development (2014) 141, 2325-2330 doi:10.1242/dev.105858 RESEARCH REPORT TECHNIQUES AND RESOURCES The tissue-specific transcriptomic landscape of the mid-gestational mouse embryo Martin Werber1, Lars Wittler1, Bernd Timmermann2, Phillip Grote1,* and Bernhard G. Herrmann1,3,* ABSTRACT mainly protein-coding genes. However, none of these datasets has Differential gene expression is a prerequisite for the formation of multiple provided an accurate representation of the transcriptome of the mouse cell types from the fertilized egg during embryogenesis. Understanding embryo. In particular, earlier studies using expression profiling did not the gene regulatory networks controlling cellular differentiation requires cover the complete set of protein coding genes or alternative transcripts the identification of crucial differentially expressed control genes and, or noncoding RNA genes. The latter have come into focus in recent ideally, the determination of the complete transcriptomes of each years, as noncoding genes are assumed to play important roles in gene individual cell type. Here, we have analyzed the transcriptomes of six regulation (for reviews, see Pauli et al., 2011; Rinn and Chang, 2012). major tissues dissected from mid-gestational (TS12) mouse embryos. Among the highly diverse class of noncoding genes, long Approximately one billion reads derived by RNA-seq analysis provided noncoding RNAs (lncRNAs) are thought to influence transcription extended transcript lengths, novel first exons and alternative transcripts by a wide range of mechanisms. For example, lncRNAs can interact of known genes. We have identified 1375 genes showing tissue-specific with chromatin-modifying protein complexes involved in gene expression, providing gene signatures for each of the six tissues. -

The Changing Chromatome As a Driver of Disease: a Panoramic View from Different Methodologies
The changing chromatome as a driver of disease: A panoramic view from different methodologies Isabel Espejo1, Luciano Di Croce,1,2,3 and Sergi Aranda1 1. Centre for Genomic Regulation (CRG), Barcelona Institute of Science and Technology, Dr. Aiguader 88, Barcelona 08003, Spain 2. Universitat Pompeu Fabra (UPF), Barcelona, Spain 3. ICREA, Pg. Lluis Companys 23, Barcelona 08010, Spain *Corresponding authors: Luciano Di Croce ([email protected]) Sergi Aranda ([email protected]) 1 GRAPHICAL ABSTRACT Chromatin-bound proteins regulate gene expression, replicate and repair DNA, and transmit epigenetic information. Several human diseases are highly influenced by alterations in the chromatin- bound proteome. Thus, biochemical approaches for the systematic characterization of the chromatome could contribute to identifying new regulators of cellular functionality, including those that are relevant to human disorders. 2 SUMMARY Chromatin-bound proteins underlie several fundamental cellular functions, such as control of gene expression and the faithful transmission of genetic and epigenetic information. Components of the chromatin proteome (the “chromatome”) are essential in human life, and mutations in chromatin-bound proteins are frequently drivers of human diseases, such as cancer. Proteomic characterization of chromatin and de novo identification of chromatin interactors could thus reveal important and perhaps unexpected players implicated in human physiology and disease. Recently, intensive research efforts have focused on developing strategies to characterize the chromatome composition. In this review, we provide an overview of the dynamic composition of the chromatome, highlight the importance of its alterations as a driving force in human disease (and particularly in cancer), and discuss the different approaches to systematically characterize the chromatin-bound proteome in a global manner. -

393LN V 393P 344SQ V 393P Probe Set Entrez Gene
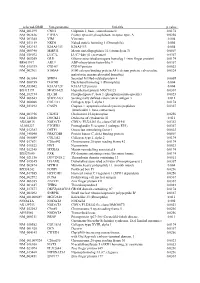
393LN v 393P 344SQ v 393P Entrez fold fold probe set Gene Gene Symbol Gene cluster Gene Title p-value change p-value change chemokine (C-C motif) ligand 21b /// chemokine (C-C motif) ligand 21a /// chemokine (C-C motif) ligand 21c 1419426_s_at 18829 /// Ccl21b /// Ccl2 1 - up 393 LN only (leucine) 0.0047 9.199837 0.45212 6.847887 nuclear factor of activated T-cells, cytoplasmic, calcineurin- 1447085_s_at 18018 Nfatc1 1 - up 393 LN only dependent 1 0.009048 12.065 0.13718 4.81 RIKEN cDNA 1453647_at 78668 9530059J11Rik1 - up 393 LN only 9530059J11 gene 0.002208 5.482897 0.27642 3.45171 transient receptor potential cation channel, subfamily 1457164_at 277328 Trpa1 1 - up 393 LN only A, member 1 0.000111 9.180344 0.01771 3.048114 regulating synaptic membrane 1422809_at 116838 Rims2 1 - up 393 LN only exocytosis 2 0.001891 8.560424 0.13159 2.980501 glial cell line derived neurotrophic factor family receptor alpha 1433716_x_at 14586 Gfra2 1 - up 393 LN only 2 0.006868 30.88736 0.01066 2.811211 1446936_at --- --- 1 - up 393 LN only --- 0.007695 6.373955 0.11733 2.480287 zinc finger protein 1438742_at 320683 Zfp629 1 - up 393 LN only 629 0.002644 5.231855 0.38124 2.377016 phospholipase A2, 1426019_at 18786 Plaa 1 - up 393 LN only activating protein 0.008657 6.2364 0.12336 2.262117 1445314_at 14009 Etv1 1 - up 393 LN only ets variant gene 1 0.007224 3.643646 0.36434 2.01989 ciliary rootlet coiled- 1427338_at 230872 Crocc 1 - up 393 LN only coil, rootletin 0.002482 7.783242 0.49977 1.794171 expressed sequence 1436585_at 99463 BB182297 1 - up 393 -

Global Patterns of Changes in the Gene Expression Associated with Genesis of Cancer a Dissertation Submitted in Partial Fulfillm
Global Patterns Of Changes In The Gene Expression Associated With Genesis Of Cancer A dissertation submitted in partial fulfillment of the requirements for the degree of Doctor of Philosophy at George Mason University By Ganiraju Manyam Master of Science IIIT-Hyderabad, 2004 Bachelor of Engineering Bharatiar University, 2002 Director: Dr. Ancha Baranova, Associate Professor Department of Molecular & Microbiology Fall Semester 2009 George Mason University Fairfax, VA Copyright: 2009 Ganiraju Manyam All Rights Reserved ii DEDICATION To my parents Pattabhi Ramanna and Veera Venkata Satyavathi who introduced me to the joy of learning. To friends, family and colleagues who have contributed in work, thought, and support to this project. iii ACKNOWLEDGEMENTS I would like to thank my advisor, Dr. Ancha Baranova, whose tolerance, patience, guidance and encouragement helped me throughout the study. This dissertation would not have been possible without her ever ending support. She is very sincere and generous with her knowledge, availability, compassion, wisdom and feedback. I would also like to thank Dr. Vikas Chandhoke for funding my research generously during my doctoral study at George Mason University. Special thanks go to Dr. Patrick Gillevet, Dr. Alessandro Giuliani, Dr. Maria Stepanova who devoted their time to provide me with their valuable contributions and guidance to formulate this project. Thanks to the faculty of Molecular and Micro Biology (MMB) department, Dr. Jim Willett and Dr. Monique Vanhoek in embedding valuable thoughts to this dissertation by being in my dissertation committee. I would also like to thank the present and previous doctoral program directors, Dr. Daniel Cox and Dr. Geraldine Grant, for facilitating, allowing, and encouraging me to work in this project. -

Mouse SNP Miner: an Annotated Database of Mouse Functional Single Nucleotide Polymorphisms Eli Reuveni*1, Vasily E Ramensky2 and Cornelius Gross1
BMC Genomics BioMed Central Database Open Access Mouse SNP Miner: an annotated database of mouse functional single nucleotide polymorphisms Eli Reuveni*1, Vasily E Ramensky2 and Cornelius Gross1 Address: 1Mouse Biology Unit, EMBL, Via Ramarini 32, 00016 Monterotondo, Italy and 2Engelhardt Institute of Molecular Biology, Vavilova 32, 119991 Moscow, Russia Email: Eli Reuveni* - [email protected]; Vasily E Ramensky - [email protected]; Cornelius Gross - [email protected] * Corresponding author Published: 21 January 2007 Received: 4 August 2006 Accepted: 21 January 2007 BMC Genomics 2007, 8:24 doi:10.1186/1471-2164-8-24 This article is available from: http://www.biomedcentral.com/1471-2164/8/24 © 2007 Reuveni et al; licensee BioMed Central Ltd. This is an Open Access article distributed under the terms of the Creative Commons Attribution License (http://creativecommons.org/licenses/by/2.0), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited. Abstract Background: The mapping of quantitative trait loci in rat and mouse has been extremely successful in identifying chromosomal regions associated with human disease-related phenotypes. However, identifying the specific phenotype-causing DNA sequence variations within a quantitative trait locus has been much more difficult. The recent availability of genomic sequence from several mouse inbred strains (including C57BL/6J, 129X1/SvJ, 129S1/SvImJ, A/J, and DBA/2J) has made it possible to catalog DNA sequence differences within a quantitative trait locus derived from crosses between these strains. However, even for well-defined quantitative trait loci (<10 Mb) the identification of candidate functional DNA sequence changes remains challenging due to the high density of sequence variation between strains. -

Investigation of Mirna‑And Lncrna‑Mediated Competing Endogenous
ONCOLOGY LETTERS 18: 5283-5293, 2019 Investigation of miRNA‑ and lncRNA‑mediated competing endogenous RNA network in cholangiocarcinoma YANXIN HE1, CHAO LIU1, PAN SONG2, ZHIGANG PANG1, ZHUOMAO MO3, CHUIGUO HUANG4, TINGTING YAN5, MENG SUN6 and XiANeN FA1 1Department of Surgery, The Second Affiliated Hospital of Zhengzhou University;2 Department of Urology, The First Affiliated Hospital of Zhengzhou University, Zhengzhou, Henan 450014; 3College of Traditional Chinese Medicine of Jinan University, Institute of Integrated Traditional Chinese and Western Medicine of Jinan University, Guangzhou, Guangdong 510632; 4Department of Urology, The Second Affiliated Hospital of Zhengzhou University, Zhengzhou, Henan 450014; 5The Nethersole School of Nursing, Faculty of Medicine, The Chinese University of Hong Kong, Hong Kong 999077; 6Department of Gynecology and Obstetrics, The Second Affiliated Hospital of Zhengzhou University, Zhengzhou, Henan 450014, P.R. China Received August 29, 2018; Accepted March 8, 2019 DOI: 10.3892/ol.2019.10852 Abstract. Cholangiocarcinoma (CCA) is a biliary malig- further evaluate the hub genes. The results obtained in the nancy which is prone to lymphatic metastasis and has a current study suggested that spalt like transcription factor 3 high mortality rate. This disease lacks effective therapeutic and OPCML intronic transcript 1 may serve an important targets and prognostic molecular biomarkers. The aim of role in the development and progression of CCA. the current study was to investigate differentially expressed genes and elucidate their association with CCA and the Introduction underlying mechanisms of action. mRNAs, long non-coding RNAs (lncRNAs) and microRNAs (miRNAs) obtained Cholangiocarcinoma (CCA) is a biliary malignancy which from 36 CCA samples and nine normal samples from The originates in the bile duct epithelium and can be subclassified Cancer Genome Atlas were integrated. -

Topological and Structural Plasticity of the Single Ig Fold and the Double Ig Fold Present in CD19
biomolecules Article Topological and Structural Plasticity of the Single Ig Fold and the Double Ig Fold Present in CD19 Philippe Youkharibache Cancer Data Science Laboratory, National Cancer Institute, NIH, Bethesda, MD 20814, USA; [email protected] Abstract: The Ig fold has had a remarkable success in vertebrate evolution, with a presence in over 2% of human genes. The Ig fold is not just the elementary structural domain of antibodies and TCRs, it is also at the heart of a staggering 30% of immunologic cell surface receptors, making it a major orchestrator of cell–cell interactions. While BCRs, TCRs, and numerous Ig-based cell surface receptors form homo- or heterodimers on the same cell surface (in cis), many of them interface as ligand-receptors (checkpoints) on interacting cells (in trans) through their Ig domains. New Ig-Ig interfaces are still being discovered between Ig-based cell surface receptors, even in well-known families such as B7. What is largely ignored, however, is that the Ig fold itself is pseudosymmetric, a property that makes the Ig domain a versatile self-associative 3D structure and may, in part, explain its success in evolution, especially through its ability to bind in cis or in trans in the context of cell surface receptor–ligand interactions. In this paper, we review the Ig domains’ tertiary and quaternary pseudosymmetries, with particular attention to the newly identified double Ig fold in the solved CD19 molecular structure to highlight the underlying fundamental folding elements of Ig domains, i.e., Ig protodomains. This pseudosymmetric property of Ig domains gives us a decoding frame of reference to understand the fold, relate all Ig domain forms, single or double, and suggest new protein engineering avenues. -
Supplementary Figures S1-S3
selected-GBID Uni-genename Uni-title p value NM_001299 CNN1 Calponin 1, basic, smooth muscle 0.0174 NM_002836 PTPRA Protein tyrosine phosphatase, receptor type, A 0.0256 NM_003380 VIM Vimentin 0.004 NM_033119 NKD1 Naked cuticle homolog 1 (Drosophila) 0.004 NM_052913 KIAA1913 KIAA1913 0.004 NM_005940 MMP11 Matrix metallopeptidase 11 (stromelysin 3) 0.0069 NM_018032 LUC7L LUC7-like (S. cerevisiae) 0.0367 NM_005269 GLI1 Glioma-associated oncogene homolog 1 (zinc finger protein) 0.0174 BE463997 ARL9 ADP-ribosylation factor-like 9 0.0367 NM_015939 CGI-09 CGI-09 protein 0.0023 NM_002961 S100A4 S100 calcium binding protein A4 (calcium protein, calvasculin, 0.0324 metastasin, murine placental homolog) NM_003014 SFRP4 Secreted frizzled-related protein 4 0.0005 NM_080759 DACH1 Dachshund homolog 1 (Drosophila) 0.004 NM_053042 KIAA1729 KIAA1729 protein 0.004 BX415194 MGC16121 Hypothetical protein MGC16121 0.0367 NM_182734 PLCB1 Phospholipase C, beta 1 (phosphoinositide-specific) 0.0023 NM_006643 SDCCAG3 Serologically defined colon cancer antigen 3 0.011 NM_000088 COL1A1 Collagen, type I, alpha 1 0.0174 NM_033292 CASP1 Caspase 1, apoptosis-related cysteine peptidase 0.0367 (interleukin 1, beta, convertase) NM_003956 CH25H Cholesterol 25-hydroxylase 0.0256 NM_144658 DOCK11 Dedicator of cytokinesis 11 0.011 AK024935 NODATA CDNA: FLJ21283 fis, clone COL01910 0.0363 AL050227 PTGER3 Prostaglandin E receptor 3 (subtype EP3) 0.0367 NM_012383 OSTF1 Osteoclast stimulating factor 1 0.0023 NM_145040 PRKCDBP Protein kinase C, delta binding protein 0.0069 NM_000089