Identification of Host Proteins Required for Vesicular Stomatitis Virus Infection
Total Page:16
File Type:pdf, Size:1020Kb

Load more
Recommended publications
-

Dissertation Philip Böhler
Three Tales of Death: New Pathways in the Induction, Inhibition and Execution of Apoptosis Inaugural-Dissertation zur Erlangung des Doktorgrades der Mathematisch-Naturwissenschaftlichen Fakultät der Heinrich-Heine-Universität Düsseldorf vorgelegt von Philip Böhler aus Bonn Düsseldorf, Juni 2019 aus dem Institut für Molekulare Medizin I der Heinrich-Heine-Universität Düsseldorf Gedruckt mit der Genehmigung der Mathematisch-Naturwissenschaftlichen Fakultät der Heinrich-Heine-Universität Düsseldorf Berichterstatter: 1. Prof. Dr. Sebastian Wesselborg 2. Prof. Dr. Henrike Heise Tag der mündlichen Prüfung: 29. Oktober 2019 “Where the first primal cell was, there was I also. Where man is, there am I. When the last life crawls under freezing stars, there will I be.” — DEATH, in: Mort, by Terry Pratchett “Right away I found out something about biology: it was very easy to find a question that was very interesting, and that nobody knew the answer to.” — Richard Feynman, in: Surely You're Joking, Mr. Feynman! Acknowledgements (Danksagung) Acknowledgements (Danksagung) Viele Menschen haben zum Gelingen meiner Forschungsarbeit und dieser Dissertation beigetragen, und nicht alle können hier namentlich erwähnt werden. Dennoch möchte ich einige besonders hervorheben. An erster Stelle möchte ich Professor Sebastian Wesselborg danken, der diese Dissertation als Erstgutachter betreut hat und der mir die Möglichkeit gab, die dazugehörigen experimentellen Arbeiten am Institut für Molekulare Medizin durchzuführen. Er und Professor Björn Stork, dem ich für die herzliche Aufnahme in seine Arbeitsgruppe danke, legten durch die richtige Mischung aus aktiver Förderung und dem Freiraum zur Umsetzung eigener wissenschaftlicher Ideen die ideale Grundlage für die Forschungsprojekte, aus denen diese Dissertation entstand. Professorin Henrike Heise, die sich freundlicherweise zur Betreuung dieser Dissertation als Zweitgutachterin bereiterklärt hat, gilt ebenfalls mein herzlicher Dank. -

Characterization of the Matrix Proteins of the Fish Rhabdovirus, Infectious Hematopoietic Necrosis Virus
AN ABSTRACT OF THE THESIS OF Patricia A. Ormonde for the degree of Master of Science presented on April 14. 1995. Title: Characterization of the Matrix Proteins of the Fish Rhabdovinis, Infectious Hematopoietic Necrosis Virus. Redacted for Privacy Abstract approved: Jo-Ann C. ong Infectious hematopoietic necrosis virus (1HNV) is an important fish pathogen enzootic in salmon and trout populations of the Pacific Northwestern United States. Occasional epizootics in fish hatcheries can result in devastating losses of fish stocks. The complete nucleotide sequence of IHNV has not yet been determined. This knowledge is the first step towards understanding the roles viral proteins play in IHNV infection, and is necessary for determining the relatedness of IHNV to other rhabdoviruses. The glycoprotein, nucleocapsid and non-virion genes of IHNV have been described previously; however, at the initiation of this study, very little was known about the matrix protein genes. Rhabdoviral matrix proteins have been found to be important in viral transcription and virion assembly. This thesis describes the preliminary characterization of the M1 and M2 matrix proteins of IHNV. In addition, the trout humoral immune response to M1 and M2 proteins expressed from plasmid DNA injected into the fish was investigated. This work may prove useful in designing future vaccines against IHN. The sequences of M1 phosphoprotein and M2 matrix protein genes of IHNV were determined from both genomic and mRNA clones. Analysis of the sequences indicated that the predicted open reading frame of M1 gene encoded a 230 amino acid protein with a estimated molecular weight of 25.6 kDa. Further analysis revealed a second open reading frame encoding a 42 amino acid protein with a calculated molecular weight of 4.8 kDa. -

Viral Strategies to Arrest Host Mrna Nuclear Export
Viruses 2013, 5, 1824-1849; doi:10.3390/v5071824 OPEN ACCESS viruses ISSN 1999-4915 www.mdpi.com/journal/viruses Review Nuclear Imprisonment: Viral Strategies to Arrest Host mRNA Nuclear Export Sharon K. Kuss *, Miguel A. Mata, Liang Zhang and Beatriz M. A. Fontoura Department of Cell Biology, University of Texas Southwestern Medical Center, Dallas, TX 75390, USA; E-Mails: [email protected] (M.A.M.); [email protected] (L.Z.); [email protected] (B.M.A.F) * Author to whom correspondence should be addressed; E-Mail: [email protected]; Tel.: +1-214-633-2001; Fax: +1-214-648-5814. Received: 10 June 2013; in revised form: 27 June 2013 / Accepted: 11 July 2013 / Published: 18 July 2013 Abstract: Viruses possess many strategies to impair host cellular responses to infection. Nuclear export of host messenger RNAs (mRNA) that encode antiviral factors is critical for antiviral protein production and control of viral infections. Several viruses have evolved sophisticated strategies to inhibit nuclear export of host mRNAs, including targeting mRNA export factors and nucleoporins to compromise their roles in nucleo-cytoplasmic trafficking of cellular mRNA. Here, we present a review of research focused on suppression of host mRNA nuclear export by viruses, including influenza A virus and vesicular stomatitis virus, and the impact of this viral suppression on host antiviral responses. Keywords: virus; influenza virus; vesicular stomatitis virus; VSV; NS1; matrix protein; nuclear export; nucleo-cytoplasmic trafficking; mRNA export; NXF1; TAP; CRM1; Rae1 1. Introduction Nucleo-cytoplasmic trafficking of proteins and RNA is critical for proper cellular functions and survival. -

Prevention & Control of Viral Hepatitis Infection
Prevention & Control of Viral Hepatitis Infection: A Strategy for Global Action © World Health Organization 2011. All rights reserved. The designations employed and the presentation of the material in this publication do not imply the expression of any opinion whatsoever on the part of the World Health Organization concerning the legal status of any country, territory, city or area or of its authorities, or concerning the delimitation of its frontiers or boundaries. Dotted lines on maps represent approximate border lines for which there may not yet be full agreement. The mention of specific companies or of certain manufacturers’ products does not imply that they are endorsed or recommended by the World Health Organization in preference to others of a similar nature that are not mentioned. Errors and omissions excepted, the names of proprietary products are distinguished by initial capital letters. All reasonable precautions have been taken by WHO to verify the information contained in this publication. However, the published material is being distributed without warranty of any kind, either express or implied. The responsibility for the interpretation and use of the material lies with the reader. In no event shall the World Health Organization be liable for damages arising from its use. Table of contents Disease burden 02 What is viral hepatitis? 05 Prevention & control: a tailored approach 06 Global Achievements 08 Remaining challenges 10 World Health Assembly: a mandate for comprehensive prevention & control 13 WHO goals and strategy -

Synthesis and Processing of the Transmembrane Envelope Protein of Equine Infectious Anemia Virus NANCY R
JOURNAL OF VIROLOGY, Aug. 1990, p. 3770-3778 Vol. 64, No. 8 0022-538X/90/083770-09$02.00/0 Copyright C 1990, American Society for Microbiology Synthesis and Processing of the Transmembrane Envelope Protein of Equine Infectious Anemia Virus NANCY R. RICE,'* LOUIS E. HENDERSON,' RAYMOND C. SOWDER,1 TERRY D. COPELAND,' STEPHEN OROSZLAN,1 AND JOHN F. EDWARDS2 Laboratory of Molecular Virology and Carcinogenesis, ABL-Basic Research Program, NCI-Frederick Cancer Research and Development Center, Frederick, Maryland 21701,1 and Department of Veterinary Pathology, College of Veterinary Medicine, Texas A&M University, College Station, Texas 778432 Received 22 December 1989/Accepted 10 May 1990 Downloaded from The transmembrane (TM) envelope protein of lentiviruses, including equine infectious anemia virus (EIAV), is significantly larger than that of other retroviruses and may extend in the C-terminal direction 100 to 200 amino acids beyond the TM domain. This size difference suggests a lentivirus-specific function for the long C-terminal extension. We have investigated the synthesis and processing of the EIAV TM protein by immune precipitation and immunoblotting experiments, by using several envelope-specific peptide antisera. We show that the TM protein in EIAV particles is cleaved by proteolysis to an N-terminal glycosylated 32- to 35-kilodalton (kDa) segment and a C-terminal nonglycosylated 20-kDa segment. The 20-kDa fragment was isolated from virus fractionated by high-pressure liquid chromatography, and its N-terminal amino acid sequence was determined for 13 residues. Together with the known nucleotide sequence, this fixes the cleavage http://jvi.asm.org/ site at a His-Leu bond located 240 amino acids from the N terminus of the TM protein. -

The Role of Hepatitis C Virus in Hepatocellular Carcinoma U
Viruses in cancer cell plasticity: the role of hepatitis C virus in hepatocellular carcinoma U. Hibner, D. Gregoire To cite this version: U. Hibner, D. Gregoire. Viruses in cancer cell plasticity: the role of hepatitis C virus in hepato- cellular carcinoma. Contemporary Oncology, Termedia Publishing House, 2015, 19 (1A), pp.A62–7. 10.5114/wo.2014.47132. hal-02187396 HAL Id: hal-02187396 https://hal.archives-ouvertes.fr/hal-02187396 Submitted on 2 Jun 2021 HAL is a multi-disciplinary open access L’archive ouverte pluridisciplinaire HAL, est archive for the deposit and dissemination of sci- destinée au dépôt et à la diffusion de documents entific research documents, whether they are pub- scientifiques de niveau recherche, publiés ou non, lished or not. The documents may come from émanant des établissements d’enseignement et de teaching and research institutions in France or recherche français ou étrangers, des laboratoires abroad, or from public or private research centers. publics ou privés. Distributed under a Creative Commons Attribution - NonCommercial - ShareAlike| 4.0 International License Review Viruses are considered as causative agents of a significant proportion of human cancers. While the very Viruses in cancer cell plasticity: stringent criteria used for their clas- sification probably lead to an under- estimation, only six human viruses the role of hepatitis C virus are currently classified as oncogenic. In this review we give a brief histor- in hepatocellular carcinoma ical account of the discovery of on- cogenic viruses and then analyse the mechanisms underlying the infectious causes of cancer. We discuss viral strategies that evolved to ensure vi- Urszula Hibner1,2,3, Damien Grégoire1,2,3 rus propagation and spread can alter cellular homeostasis in a way that increases the probability of oncogen- 1Institut de Génétique Moléculaire de Montpellier, CNRS, UMR 5535, Montpellier, France ic transformation and acquisition of 2Université Montpellier 2, Montpellier, France stem cell phenotype. -

A Computational Approach for Defining a Signature of Β-Cell Golgi Stress in Diabetes Mellitus
Page 1 of 781 Diabetes A Computational Approach for Defining a Signature of β-Cell Golgi Stress in Diabetes Mellitus Robert N. Bone1,6,7, Olufunmilola Oyebamiji2, Sayali Talware2, Sharmila Selvaraj2, Preethi Krishnan3,6, Farooq Syed1,6,7, Huanmei Wu2, Carmella Evans-Molina 1,3,4,5,6,7,8* Departments of 1Pediatrics, 3Medicine, 4Anatomy, Cell Biology & Physiology, 5Biochemistry & Molecular Biology, the 6Center for Diabetes & Metabolic Diseases, and the 7Herman B. Wells Center for Pediatric Research, Indiana University School of Medicine, Indianapolis, IN 46202; 2Department of BioHealth Informatics, Indiana University-Purdue University Indianapolis, Indianapolis, IN, 46202; 8Roudebush VA Medical Center, Indianapolis, IN 46202. *Corresponding Author(s): Carmella Evans-Molina, MD, PhD ([email protected]) Indiana University School of Medicine, 635 Barnhill Drive, MS 2031A, Indianapolis, IN 46202, Telephone: (317) 274-4145, Fax (317) 274-4107 Running Title: Golgi Stress Response in Diabetes Word Count: 4358 Number of Figures: 6 Keywords: Golgi apparatus stress, Islets, β cell, Type 1 diabetes, Type 2 diabetes 1 Diabetes Publish Ahead of Print, published online August 20, 2020 Diabetes Page 2 of 781 ABSTRACT The Golgi apparatus (GA) is an important site of insulin processing and granule maturation, but whether GA organelle dysfunction and GA stress are present in the diabetic β-cell has not been tested. We utilized an informatics-based approach to develop a transcriptional signature of β-cell GA stress using existing RNA sequencing and microarray datasets generated using human islets from donors with diabetes and islets where type 1(T1D) and type 2 diabetes (T2D) had been modeled ex vivo. To narrow our results to GA-specific genes, we applied a filter set of 1,030 genes accepted as GA associated. -

4-6 Weeks Old Female C57BL/6 Mice Obtained from Jackson Labs Were Used for Cell Isolation
Methods Mice: 4-6 weeks old female C57BL/6 mice obtained from Jackson labs were used for cell isolation. Female Foxp3-IRES-GFP reporter mice (1), backcrossed to B6/C57 background for 10 generations, were used for the isolation of naïve CD4 and naïve CD8 cells for the RNAseq experiments. The mice were housed in pathogen-free animal facility in the La Jolla Institute for Allergy and Immunology and were used according to protocols approved by the Institutional Animal Care and use Committee. Preparation of cells: Subsets of thymocytes were isolated by cell sorting as previously described (2), after cell surface staining using CD4 (GK1.5), CD8 (53-6.7), CD3ε (145- 2C11), CD24 (M1/69) (all from Biolegend). DP cells: CD4+CD8 int/hi; CD4 SP cells: CD4CD3 hi, CD24 int/lo; CD8 SP cells: CD8 int/hi CD4 CD3 hi, CD24 int/lo (Fig S2). Peripheral subsets were isolated after pooling spleen and lymph nodes. T cells were enriched by negative isolation using Dynabeads (Dynabeads untouched mouse T cells, 11413D, Invitrogen). After surface staining for CD4 (GK1.5), CD8 (53-6.7), CD62L (MEL-14), CD25 (PC61) and CD44 (IM7), naïve CD4+CD62L hiCD25-CD44lo and naïve CD8+CD62L hiCD25-CD44lo were obtained by sorting (BD FACS Aria). Additionally, for the RNAseq experiments, CD4 and CD8 naïve cells were isolated by sorting T cells from the Foxp3- IRES-GFP mice: CD4+CD62LhiCD25–CD44lo GFP(FOXP3)– and CD8+CD62LhiCD25– CD44lo GFP(FOXP3)– (antibodies were from Biolegend). In some cases, naïve CD4 cells were cultured in vitro under Th1 or Th2 polarizing conditions (3, 4). -

Tricarboxylic Acid Cycle Metabolites As Mediators of DNA Methylation Reprogramming in Bovine Preimplantation Embryos
Supplementary Materials Tricarboxylic Acid Cycle Metabolites as Mediators of DNA Methylation Reprogramming in Bovine Preimplantation Embryos Figure S1. (A) Total number of cells in fast (FBL) and slow (SBL) blastocysts; (B) Fluorescence intensity for 5-methylcytosine and 5-hydroxymethylcytosine of fast and slow blastocysts of cells from Trophoectoderm (TE) or inner cell mass (ICM). Fluorescence intensity for 5-methylcytosine of cells from the ICM or TE in blastocysts cultured with (C) dimethyl-succinate or (D) dimethyl-α- ketoglutarate. Statistical significance is identified by different letters. Figure S2. Experimental design. Table S1. Selected genes related to metabolism and epigenetic mechanisms from RNA-Seq analysis of bovine blastocysts (slow vs. fast). Genes in blue represent upregulation in slow blastocysts, genes in red represent upregulation in fast blastocysts. log2FoldCh Gene p-value p-Adj ange PDHB −1.425 0.000 0.000 MDH1 −1.206 0.000 0.000 APEX1 −1.193 0.000 0.000 OGDHL −3.417 0.000 0.002 PGK1 −0.942 0.000 0.002 GLS2 1.493 0.000 0.002 AICDA 1.171 0.001 0.005 ACO2 0.693 0.002 0.011 CS −0.660 0.002 0.011 SLC25A1 1.181 0.007 0.032 IDH3A −0.728 0.008 0.035 GSS 1.039 0.013 0.053 TET3 0.662 0.026 0.093 GLUD1 −0.450 0.032 0.108 SDHD −0.619 0.049 0.143 FH −0.547 0.054 0.149 OGDH 0.316 0.133 0.287 ACO1 −0.364 0.141 0.297 SDHC −0.335 0.149 0.311 LIG3 0.338 0.165 0.334 SUCLG −0.332 0.174 0.349 SDHA 0.297 0.210 0.396 SUCLA2 −0.324 0.248 0.439 DNMT1 0.266 0.279 0.486 IDH3B1 −0.269 0.296 0.503 SDHB −0.213 0.339 0.544 DNMT3B 0.181 0.386 0.598 APOBEC1 0.629 0.386 0.598 TDG 0.427 0.398 0.611 IDH3G 0.237 0.468 0.675 NEIL2 0.509 0.572 0.720 IDH2 0.298 0.571 0.720 DNMT3L 1.306 0.590 0.722 GLS 0.120 0.706 0.821 XRCC1 0.108 0.793 0.887 TET1 −0.028 0.879 0.919 DNMT3A 0.029 0.893 0.920 MBD4 −0.056 0.885 0.920 PDHX 0.033 0.890 0.920 SMUG1 0.053 0.936 0.954 TET2 −0.002 0.991 0.991 Table S2. -

Abstract Book
Table of Contents Tuesday, May 25, 2021 ............................................................................................................................................................... 5 T1. Astrocyte-Specific Expression of the Extracellular Matrix Gene HtrA1 Regulates Susceptibility to Stress in a Sex- Specific Manner ....................................................................................................................................................................... 5 T2. Plexin-B2 Regulates Migratory Plasticity of Glioblastoma Cells in a 3D-Printed Micropattern Device ............................. 5 T3. Pathoanatomical Mapping of Differential MAPT Expression and Splicing in Progressive Supranuclear Palsy ............... 5 T4. Behavioral Variability in Response to Chronic Stress and Morphine in BXD and Parental Mouse Lines......................... 6 T5. Thyroid-Stimulating Hormone Receptor Regulates Anxiety .............................................................................................. 6 T6. Drugs That Inhibit Microglial Inflammation Also Ameliorate Aβ1-42 Induced Toxicity in C. Elegans ............................... 7 T7. Phosphodiesterase 1b is an Upstream Regulator of a Key Gene Network in the Nucleus Accumbens Driving Addiction- Like Behaviors ......................................................................................................................................................................... 7 T8. Reduced Gap Effect in Children With FOXP1 Syndrome and Autism Spectrum -

Rational Engineering of HCV Vaccines for Humoral Immunity
viruses Review To Include or Occlude: Rational Engineering of HCV Vaccines for Humoral Immunity Felicia Schlotthauer 1,2,†, Joey McGregor 1,2,† and Heidi E Drummer 1,2,3,* 1 Viral Entry and Vaccines Group, Burnet Institute, Melbourne, VIC 3004, Australia; [email protected] (F.S.); [email protected] (J.M.) 2 Department of Microbiology and Immunology, Peter Doherty Institute for Infection and Immunity, University of Melbourne, Melbourne, VIC 3000, Australia 3 Department of Microbiology, Monash University, Clayton, VIC 3800, Australia * Correspondence: [email protected]; Tel.: +61-392-822-179 † These authors contributed equally to this work. Abstract: Direct-acting antiviral agents have proven highly effective at treating existing hepatitis C infections but despite their availability most countries will not reach the World Health Organization targets for elimination of HCV by 2030. A prophylactic vaccine remains a high priority. Whilst early vaccines focused largely on generating T cell immunity, attention is now aimed at vaccines that gen- erate humoral immunity, either alone or in combination with T cell-based vaccines. High-resolution structures of hepatitis C viral glycoproteins and their interaction with monoclonal antibodies isolated from both cleared and chronically infected people, together with advances in vaccine technologies, provide new avenues for vaccine development. Keywords: glycoprotein E2; vaccine development; humoral immunity Citation: Schlotthauer, F.; McGregor, J.; Drummer, H.E To Include or 1. Introduction Occlude: Rational Engineering of HCV Vaccines for Humoral Immunity. The development of direct-acting antiviral agents (DAA) with their ability to cure Viruses 2021, 13, 805. https:// infection in >95% of those treated was heralded as the key to eliminating hepatitis C globally. -
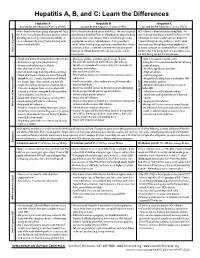
Hepatitis A, B, and C: Learn the Differences
Hepatitis A, B, and C: Learn the Differences Hepatitis A Hepatitis B Hepatitis C caused by the hepatitis A virus (HAV) caused by the hepatitis B virus (HBV) caused by the hepatitis C virus (HCV) HAV is found in the feces (poop) of people with hepa- HBV is found in blood and certain body fluids. The virus is spread HCV is found in blood and certain body fluids. The titis A and is usually spread by close personal contact when blood or body fluid from an infected person enters the body virus is spread when blood or body fluid from an HCV- (including sex or living in the same household). It of a person who is not immune. HBV is spread through having infected person enters another person’s body. HCV can also be spread by eating food or drinking water unprotected sex with an infected person, sharing needles or is spread through sharing needles or “works” when contaminated with HAV. “works” when shooting drugs, exposure to needlesticks or sharps shooting drugs, through exposure to needlesticks on the job, or from an infected mother to her baby during birth. or sharps on the job, or sometimes from an infected How is it spread? Exposure to infected blood in ANY situation can be a risk for mother to her baby during birth. It is possible to trans- transmission. mit HCV during sex, but it is not common. • People who wish to be protected from HAV infection • All infants, children, and teens ages 0 through 18 years There is no vaccine to prevent HCV.