Theory of Linear Sweep Voltammetry with Diffuse Charge: Unsupported Electrolytes, Thin films, and Leaky Membranes
Total Page:16
File Type:pdf, Size:1020Kb

Load more
Recommended publications
-

07 Chapter2.Pdf
22 METHODOLOGY 2.1 INTRODUCTION TO ELECTROCHEMICAL TECHNIQUES Electrochemical techniques of analysis involve the measurement of voltage or current. Such methods are concerned with the interplay between solution/electrode interfaces. The methods involve the changes of current, potential and charge as a function of chemical reactions. One or more of the four parameters i.e. potential, current, charge and time can be measured in these techniques and by plotting the graphs of these different parameters in various ways, one can get the desired information. Sensitivity, short analysis time, wide range of temperature, simplicity, use of many solvents are some of the advantages of these methods over the others which makes them useful in kinetic and thermodynamic studies1-3. In general, three electrodes viz., working electrode, the reference electrode, and the counter or auxiliary electrode are used for the measurement in electrochemical techniques. Depending on the combinations of parameters and types of electrodes there are various electrochemical techniques. These include potentiometry, polarography, voltammetry, cyclic voltammetry, chronopotentiometry, linear sweep techniques, amperometry, pulsed techniques etc. These techniques are mainly classified into static and dynamic methods. Static methods are those in which no current passes through the electrode-solution interface and the concentration of analyte species remains constant as in potentiometry. In dynamic methods, a current flows across the electrode-solution interface and the concentration of species changes such as in voltammetry and coulometry4. 2.2 VOLTAMMETRY The field of voltammetry was developed from polarography, which was invented by the Czechoslovakian Chemist Jaroslav Heyrovsky in the early 1920s5. Voltammetry is an electrochemical technique of analysis which includes the measurement of current as a function of applied potential under the conditions that promote polarization of working electrode6. -

Hydrodynamic Voltammetry As a Rapid and Simple Method for Evaluating Soil Enzyme Activities
Sensors 2015, 15, 5331-5343; doi:10.3390/s150305331 OPEN ACCESS sensors ISSN 1424-8220 www.mdpi.com/journal/sensors Article Hydrodynamic Voltammetry as a Rapid and Simple Method for Evaluating Soil Enzyme Activities Kazuto Sazawa 1,* and Hideki Kuramitz 2 1 Center for Far Eastern Studies, University of Toyama, Gofuku 3190, 930-8555 Toyama, Japan 2 Department of Environmental Biology and Chemistry, Graduate School of Science and Engineering for Research, University of Toyama, Gofuku 3190, 930-8555 Toyama, Japan; E-Mail: [email protected] * Author to whom correspondence should be addressed; E-Mail: [email protected]; Tel./Fax: +81-76-445-66-69. Academic Editor: Ki-Hyun Kim Received: 26 December 2014 / Accepted: 28 February 2015 / Published: 4 March 2015 Abstract: Soil enzymes play essential roles in catalyzing reactions necessary for nutrient cycling in the biosphere. They are also sensitive indicators of ecosystem stress, therefore their evaluation is very important in assessing soil health and quality. The standard soil enzyme assay method based on spectroscopic detection is a complicated operation that requires the removal of soil particles. The purpose of this study was to develop a new soil enzyme assay based on hydrodynamic electrochemical detection using a rotating disk electrode in a microliter droplet. The activities of enzymes were determined by measuring the electrochemical oxidation of p-aminophenol (PAP), following the enzymatic conversion of substrate-conjugated PAP. The calibration curves of β-galactosidase (β-gal), β-glucosidase (β-glu) and acid phosphatase (AcP) showed good linear correlation after being spiked in soils using chronoamperometry. -

Linear Sweep Voltammetric Determination of Free Chlorine in Waters Using Graphite Working Electrodes
J.Natn.Sci.FoundationLinear sweep voltammetric Sri Lanka determination 2008 36 (1):of free 25-31 chlorine 25 RESEARCH ARTICLE Linear sweep voltammetric determination of free chlorine in waters using graphite working electrodes K.A.S. Pathiratne*, S.S. Skandaraja and E.M.C.M. Jayasena Department of Chemistry, Faculty of Science, University of Kelaniya, Kelaniya. Revised: 16 June 2007 ; Accepted: 23 July 2007 Abstract: Applicability of linear sweep voltammetry using and stored. During disinfecting processes, it is added graphite working electrodes for determination of free chlorine in to potable waters and it undergoes hydrolysis in water waters was demonstrated. Influence of the nature of supporting forming hypochlorous acid and hypochlorous ions. electrolyte, its concentrations, pH and rate of potential variation of working electrode on voltammetric responses corresponding - NaOCl + H O HOCl + NaOH (1) to the oxidation of ClO were examined. It was found that, any 2 of the salt solutions KNO , K SO or Na SO at the optimum 3 2 4 2 4 Kd concentration of 0.1 mol dm-3 could be used as a supporting HOCl ClO- + H+ (2) electrolyte for the above determination. The study also revealed (Dissociation Constant, K = 2.9 x 10-8 mol dm-3) that, any pH in the range of 8.5 to 11 could yield satisfactory d results. The anodic peak current at the working electrode potential of +1.030 V vs Ag/AgCl reference electrode was found As shown by equation (2), hypochlorous acid to linearly increase with concentration of free chlorine up to undergoes further dissociation and depending on the pH, 300 mg dm-3 (R2 = 0.9996). -

Linear Sweep Anodic Stripping Voltammetry: Determination of Chromium (VI) Using Synthesized Gold Nanoparticles Modified Screen-Printed Electrode
J. Chem. Sci. Vol. 127, No. 6, June 2015, pp. 1075–1081. c Indian Academy of Sciences. DOI 10.1007/s12039-015-0864-4 Linear sweep anodic stripping voltammetry: Determination of Chromium (VI) using synthesized gold nanoparticles modified screen-printed electrode SALAMATU ALIYU TUKURa,b, NOR AZAH YUSOFa,c,∗ and REZA HAJIANc,∗ aDepartment of Chemistry, Faculty of Science, Universiti Putra Malaysia, 43400 UPM, Serdang, Selangor, Malaysia bDepartment of Chemistry, Faculty of Science, Kaduna State University, Kaduna, Nigeria cInstitute of Advanced Technology, Universiti Putra Malaysia, Serdang, Selangor 43400, Malaysia e-mail: [email protected]; [email protected] MS received 16 October 2014; revised 17 February 2015; accepted 19 February 2015 Abstract. A highly sensitive electrochemical sensor has been constructed for determination of Cr(VI) with the lowest limit of detection (LOD) reported to date using gold nanoparticles (AuNPs) modified screen-printed electrode (SPE). The modification of SPE by casting pure AuNPs increases the sensitivity for detection of Cr(VI) ion using anodic stripping voltammetry. Cr(VI) ions are reduced to chromium metal on SPE-AuNPs by applying deposition potential of –1.1 V for 180 s. Afterwards, the oxidation peak current of chromium is obtained by linear sweep voltammetry in the range of −1.0 V to 0.2 V. Under the optimized conditions (HClO4, 0.06 mol L−1; deposition potential, –1.1 V; deposition time, 180s; scan rate, 0.1 V s−1), the limit of detection (LOD) was 1.6 pg mL−1. The fabricated electrode was successfully used for detection of Cr(VI) in tap and seawater. -

Cyclic Voltammetry
Cyclic Voltammetry Denis Andrienko January 22, 2008 2 Literature: 1. Allen J. Bard, Larry R. Faulkner “Electrochemical Methods: Fundamentals and Applications” 2. http://www.cheng.cam.ac.uk/research/groups/electrochem/teaching.html Chapter 1 Cyclic Voltammetry 1.1 Background Cyclic voltammetry is the most widely used technique for acquiring qualitative information about elec- trochemical reactions. it offers a rapid location of redox potentials of the electroactive species. A few concepts has to be introduced before talking about this method. 1.1.1 Electronegativity Electronegativity is the affinity for electrons. The atoms of the various elements differ in their affinity for electrons. The term was first proposed by Linus Pauling in 1932 as a development of valence bond theory. The table for all elements can be looked up on Wikipedia: http://en.wikipedia.org/wiki/Electronegativity. Some facts to remember: • Fluorine (F) is the most electronegative element. χF = 3.98. • The electronegativity of oxygen (O) χO = 3.44 is exploited by life, via shuttling of electrons between carbon (C, χF = 2.55) and oxygen (O): Moving electrons against the gradient (O to C) - as occurs in photosynthesis - requires energy (and stores it). Moving electrons down the gradient (C to O) - as occurs in cellular respiration - releases energy. • The relative electronegativity of two interacting atoms plays a major part in determining what kind of chemical bond forms between them. Examples: • Sodium (χNa = 0.93) and Chlorine (χCl = 3.16) = Ionic Bond: There is a large difference in electronegativity, so the chlorine atom takes an electron from the sodium atom converting the atoms into ions (Na+) and (Cl−). -

Waveneuro 4 Fast-Scan Cyclic Voltammetry (FSCV) Potentiostat System
WaveNeuro 4 Fast-Scan Cyclic Voltammetry (FSCV) Potentiostat System Pine Research Instrumentation 2741 Campus Walk Avenue Building 100 Durham, NC 27705 USA http://www.pineresearch.com Phone: +1 919.782.8320 Fax: +1 919.782.8323 Copyright © 2008 - 2019 Pine Research Instrumentation DRU10227 (REV001 / FEB 2019) Table of Contents 1. PREFACE 1 1.1 Scope .................................................................................................................................... 1 1.2 Copyright .............................................................................................................................. 1 1.3 Trademarks ........................................................................................................................... 1 1.4 Use Limitation ....................................................................................................................... 1 1.5 Service and Warranty Information .................................................................................... 2 1.6 Icons (Icônes) ....................................................................................................................... 3 1.7 Back Panel Markings ........................................................................................................... 4 1.7.1 Serial Number ............................................................................................................................. 4 1.7.2 Model Numbers ......................................................................................................................... -

For Peer Review Only
IUPAC Terminology of electrochemical methods of analysis Journal: Pure and Applied Chemistry ManuscriptFor ID PeerPAC-REC-18-01-09.R2 Review Only Manuscript Type: Recommendation Date Submitted by the 27-Feb-2019 Author: Complete List of Authors: Pingarron, José; Complutense University of Madrid, Department of Analytical Chemistry Labuda, Jan; Slovak University of Technology, Department of Analytical Chemistry Barek, Jiří ; Charles University, Faculty of Science, Department of Analytical Chemistry Brett, Christopher; Universidade de Coimbra, Departamento de Química Camões, Maria; Universidade de Lisboa, Centro de Química Estrutural Fojta, Miroslav; Akademie ved Ceské Republiky, Institute of Biophysics Hibbert, David; University of New South Wales, School of Chemistry electoanalytical chemistry, electrodes, potentiometry, amperometry, Keywords: voltammetry, coulometry, electrogravimetry, conductometry, impedimetry, spectroelectrochemistry Author-Supplied Keywords: P.O. 13757, Research Triangle Park, NC (919) 485-8700 Page 1 of 62 IUPAC 1 2 3 4 IUPAC Provisional Recommendation 5 6 7 José M. Pingarrón1, Ján Labuda2, Jiří Barek3, Christopher M.A. Brett4, Maria Filomena 8 Camões5, Miroslav Fojta6, D. Brynn Hibbert7*. 9 10 11 Terminology of electrochemical methods of analysis (IUPAC 12 Recommendations 201x) 13 14 15 16 Abstract: Recommendations are given concerning the terminology of methods used in 17 electroanalytical chemistry.For PeerFundamental Review terms in electrochemistry Only are reproduced from 18 previous PAC Recommendations, and -
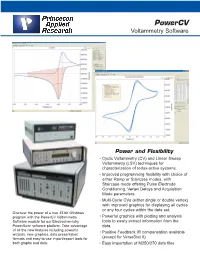
Powercv Voltammetry Software
PowerCV Voltammetry Software Power and Flexibility • Cyclic Voltammetry (CV) and Linear Sweep Voltammetry (LSV) techniques for characterization of redox-active systems. • Improved programming flexibility with choice of either Ramp or Staircase modes, with Staircase mode offering Pulse Electrode Conditioning, Vertex Delays and Acquisition Mode parameters. • Multi-Cycle CVs (either single or double vertex) with improved graphics for displaying all cycles or any four cycles within the data set. Discover the power of a true 32-bit Windows program with the PowerCV Voltammetry • Powerful graphics with plotting and analysis Software module for our Electrochemistry tools to easily extract information from the PowerSuite software platform. Take advantage data. of all the new features including powerful • Positive Feedback IR compensation available wizards, new graphics, data presentation formats and easy-to-use import/export tools for (except for VersaStat II) both graphs and data. • Easy importation of M250/270 data files Discover the Power in PowerCV 32 PowerCV continues our long tradition of providing the leading voltammetry software to the research electrochemistry community. PowerCV, the PowerSuite module that offers Cyclic Voltammetry (CV) and Linear Sweep Volatmmetry (LSV) techniques for data acquisition and analysis with Princeton Applied Research systems, has expanded its capabilities further with the addition of new waveform programming options and analysis tools. LSV and CV are the most widely used techniques in electrochemical studies as they provide a fast and simple way to characterize a redox-active system. In these techniques, the potentiostat applies a linear sweep of a programmed potential range and monitors the current as a function of the applied potential. -

Electrochemical Methods of Analysis Thomas Wenzel Department of Chemistry Bates College, Lewiston ME 04240 [email protected]
Electrochemical Methods of Analysis Thomas Wenzel Department of Chemistry Bates College, Lewiston ME 04240 [email protected] The following textual material is designed to accompany a series of in-class problem sets that develop many of the fundamental aspects of electrochemical analytical methods. TABLE OF CONTENTS 1. Basic Concepts in Electrochemistry 2 2. The Chemical Energy of a System 4 3. Relationship of Chemical Energy to Electrochemical Potential 10 4. Table of Standard State Electrochemical Potentials 12 5. Electrochemical Cells 14 6. Potential of an Electrochemical Cell 20 7. Electrochemical Analytical Methods 24 7.1. Ion-Selective Electrodes 25 7.1.1. pH Electrode 25 7.1.2. Other Glass Electrodes 27 7.1.3. Membrane Electrodes 27 7.1.4. Enzyme Electrodes 28 7.1.5. Solid-State Electrodes 28 7.1.6. Gas-Sensing Electrodes 28 7.2. Electrodeposition/Electrogravimetry 29 7.3. Coulometry 31 7.4. Titrimetric Methods of Analysis 33 7.4.1. “Classical” Redox Titration 33 7.4.2. Coulometric Titration (Controlled Current Coulometry) 33 7.4.3. Amperometric Titration 35 7.4.4. Potentiometric Titration 37 7.5. Voltammetric Methods 44 7.5.1. Anodic Stripping Voltammetry 46 7.5.2. Linear Sweep Voltammetry 49 7.5.3. Differential Pulse Linear Sweep Voltammetry 52 7.5.4. Cyclic Voltammetry 55 1 1. Basic Concepts in Electrochemistry Electrochemical processes are commonly used for analytical measurements. There are a variety of electrochemical methods with different degrees of utility for quantitative and qualitative analysis that are included in this unit. The coverage herein is not exhaustive and methods that are most important or demonstrate different aspects of electrochemical measurements are included. -

How to Cite Complete Issue More Information About This
Revista Cubana de Química ISSN: 0258-5995 ISSN: 2224-5421 Universidad de Oriente Vilasó-Cadre, Javier Ernesto; Arada-Pérez, María de los Ángeles; Arce-Castro, Jorge; Baeza-Reyes, Dr.C. Study of the potential sweeps in the manual staircase voltammetry Revista Cubana de Química, vol. 32, no. 2, 2020, May-August, pp. 287-298 Universidad de Oriente Available in: http://www.redalyc.org/articulo.oa?id=443564573008 How to cite Complete issue Scientific Information System Redalyc More information about this article Network of Scientific Journals from Latin America and the Caribbean, Spain and Journal's webpage in redalyc.org Portugal Project academic non-profit, developed under the open access initiative Rev. Cubana Quím. Vol.32, no.2 mayo-agosto, 2020, págs. 287-298, e-ISSN: 2224-5421 Artículo original Study of the potential sweeps in the manual staircase voltammetry Estudio de los barridos de potencial en la voltamperometría manual de barrido en escalera MSc. Javier Ernesto Vilasó-Cadre1* https://orcid.org/0000-0001-5172-7136 Dra.C. María de los Ángeles Arada-Pérez2 https://orcid.org/0000-0001-9262-2066 Jorge Arce-Castro2 https://orcid.org/0000-0002-5443-6796 Dr.C. José Alejandro Baeza-Reyes3 https://orcid.org/0000-0001-9621-8175 1Institute of Metallurgy. Universidad Autónoma de San Luis Potosí, México. 2Faculty of Natural and Exact Sciences. Universidad de Oriente, Santiago de Cuba, Cuba. 3Faculty of Chemistry. Universidad Nacional Autónoma de México. *Autor para la correspondencia. correo electrónico: [email protected] ABSTRACT Voltammetry can be developed using manual potentiostats. This study focuses on the potential sweeps and its influence on the voltammograms in the manual staircase voltammetry. -

Analytical Methods Accepted Manuscript
Analytical Methods Accepted Manuscript This is an Accepted Manuscript, which has been through the Royal Society of Chemistry peer review process and has been accepted for publication. Accepted Manuscripts are published online shortly after acceptance, before technical editing, formatting and proof reading. Using this free service, authors can make their results available to the community, in citable form, before we publish the edited article. We will replace this Accepted Manuscript with the edited and formatted Advance Article as soon as it is available. You can find more information about Accepted Manuscripts in the Information for Authors. Please note that technical editing may introduce minor changes to the text and/or graphics, which may alter content. The journal’s standard Terms & Conditions and the Ethical guidelines still apply. In no event shall the Royal Society of Chemistry be held responsible for any errors or omissions in this Accepted Manuscript or any consequences arising from the use of any information it contains. www.rsc.org/methods Page 1 of 22 Analytical Methods 1 2 3 4 The Voltammetric Behaviour of Lead at a Hand Drawn 5 Pencil Electrode and Its Trace Determination in Water by 6 7 Stripping Voltammetry 8 9 10 Kevin C. Honeychurch 11 12 Department of Biological, Biomedical and Analytical Sciences, Faculty of Applied Sciences, 13 University of the West of England, Bristol, UK 14 15 Corresponding author Tel.: +44 117 3282977. Fax: +44 117 3282904 Email: 16 17 [email protected] 18 19 Abstract 20 21 This is the first reported the proof of concept application of pencil drawn electrodes (PDEs) for anodic Manuscript 22 23 stripping voltammetric determination of Pb 2+ or any metal ion. -

Autolab Brochure 2013 EN LR.Pdf
Metrohm Autolab Instruments for electrochemical research 02 Metrohm Autolab • Founded in 1986 • Based in Utrecht, The Netherlands • Since 1999 part of the Metrohm Group • Introduced the first computer controlled potentiostat/galvanostat • Develops and produces the high quality Autolab range of products • Strong background in electrochemistry • Supported by the worldwide Metrohm distribution network • Three years factory warranty on all instruments • Dedicated to research International presence Eco Chemie – Metrohm Autolab Eco Chemie was founded in 1986 and is since 1999 a Metrohm Autolab based in Utrecht, The Netherlands, 03 member of the Metrohm group of companies. In 2009 designs and manufactures Autolab instruments, acces- the company name changed to Metrohm Autolab to sories, and software for electrochemistry. reflect the customer oriented combination of the world- wide Metrohm sales and support organization and the Known for innovation, the Autolab was the first commer high quality Autolab series of instruments developed by cial digital potentiostat/galvanostat, that was completely Eco Chemie. computer controlled. Our latest software package NOVA has again set a high standard for powerful electrochemi- Metrohm Autolab is an ISO9001 certified company. cal research software. With our background and knowledge in electrochemistry and our worldwide distributor network, our mission is to serve the research community all over the world by supplying state of the art instruments and unrivalled support. All Metrohm Autolab instruments are covered by a three year factory warranty. 04 Metrohm Autolab – part of the global Metrohm Group Metrohm Autolab is a member of the global Metrohm Autolab products can be combined easily with many Group. Since the first days of the Autolab company, Metrohm instruments, notably for automation.