2.0 Mg Film-Coated Tablets 2
Total Page:16
File Type:pdf, Size:1020Kb

Load more
Recommended publications
-

Cyproterone Art. 31
Приложение I Списък на лекарствените продукти и форми 1 Държава членка Притежател на Наименование на INN/Активно Фармацевтична Начин на (ЕИП) разрешението за продукта вещество + форма приложение употреба Количество на активното вещество (в дозова единица) Австрия Bayer Austria Gmbh Androcur Depot Cyproterone Acetate Инжекционен Интрамускулно 300mg/3ml разтвор приложение Австрия Bayer Austria Gmbh Climen Cyproterone Acetate 1mg Обвита таблетка Перорално таблетка, Estradiol приложение Valerate 2mg таблетка| Estradiol Valerate 2mg таблетка Австрия Bayer Austria Gmbh Climen 28-Tage Cyproterone Acetate 1mg Обвита таблетка Перорално таблетка, Estradiol приложение Valerate 2mg таблетка| Estradiol Valerate 2mg таблетка Австрия Bayer Austria Gmbh Diane Mite Cyproterone Acetate 2mg Обвита таблетка Перорално таблетка, Ethinylestradiol приложение 35μg таблетка Австрия Bayer Austria Gmbh Minerva Cyproterone Acetate 2mg Обвита таблетка Перорално таблетка, Ethinylestradiol приложение 0,035mg таблетка Австрия Bayer Austria Gmbh Andro-Diane Cyproterone Acetate Таблетка Перорално 10mg таблетка приложение Австрия Bayer Austria Gmbh Androcur Cyproterone Acetate Таблетка Перорално 100mg таблетка приложение Австрия Bayer Austria Gmbh Androcur Cyproterone Acetate Таблетка Перорално 50mg таблетка приложение Австрия Gynial Gmbh Alisma Cyproterone Acetate 2mg Филмирана таблетка Перорално таблетка, Ethinylestradiol приложение 35μg таблетка 2 Държава членка Притежател на Наименование на INN/Активно Фармацевтична Начин на (ЕИП) разрешението за продукта вещество + -

Aldosterone Enhances Renal Calcium Reabsorption by Two Types of Channels
View metadata, citation and similar papers at core.ac.uk brought to you by CORE provided by Elsevier - Publisher Connector Kidney International, Vol. 66 (2004), pp. 242–250 Aldosterone enhances renal calcium reabsorption by two types of channels MARIE LECLERC,MICHELE` G. BRUNETTE, and DENIS COUCHOUREL Maisonneuve-Rosemont Hospital and University of Montreal, Canada Aldosterone enhances renal calcium reabsorption by two types transepithelial potential difference in the late segments of channels. of the nephron, reflecting a stimulation of the amiloride- Background. Aldosterone has been known for many years + + sensitive Na transport by the apical membranes. to increase sodium (Na ) reabsorption by the distal nephron. The present in vitro experiments investigated the effect of the In contrast to the abundant literature dealing with the + hormone on calcium (Ca2 ) transport by the luminal membrane antinatriuretic effect of aldosterone, data concerning its + of the rabbit nephron, independent of any systemic influence. action on Ca2 transport are relatively scarce. Clearance Methods. Proximal and distal tubules were incubated with studies showed either an increase in calciuria [2] or an + either aldosterone or the carrier. The luminal membranes of absence of significant change of Ca2 excretion [3, 4] af- these tubules were purified, vesiculated, and 45Ca uptake by these vesicles was subsequently measured. ter aldosterone administration. In a randomized study in Results. Treatment of the distal tubules with 10−8 mol/L al- male volunteers, Van Hamersvelt et al [5] compared the + + dosterone enhanced both 0.1 and 0.5 mmol/L Ca2 transport. effect of the Ca2 channel blocker felodipine, alone or The hormone action was abolished by tyrosine kinase inhibitors. -
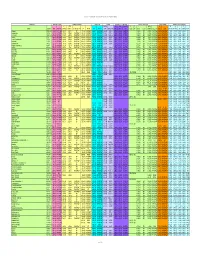
Chemical-Specific Parameters Supporting Table May 2016 Analyte
Regional Screening Level (RSL) Chemical-specific Parameters Supporting Table May 2016 Contaminant Molecular Weight Volatility Parameters Melting Point Density Diffusivity in Air and Water Partition Coefficients Water Solubility Tapwater Dermal Parameters H` (atm- Density Dia Diw Dia and Diw Kd Kd Koc log Kow S B τevent t* Kp 3 3 2 2 Analyte CAS No. MW MW Ref (unitless) m /mole) H` and HLC Ref VP VP Ref MP MP Ref (g/cm ) Density Ref (cm /s) (cm /s) Ref (L/kg) Ref (L/kg) Koc Ref (unitless) log Kow Ref (mg/L) S Ref (unitless) (hr/event) (hr) (cm/hr) K Ref Acephate 30560-19-1 1.8E+02 PHYSPRO 2.0E-11 5.0E-13 EPI 1.7E-06 PHYSPROP 8.8E+01 PHYSPROP 1.4E+00 CRC89 3.7E-02 8.0E-06 WATER9 1.0E+01 EPI -8.5E-01 PHYSPRO 8.2E+05 PHYSPROP 2.1E-04 1.1E+00 2.7E+00 4.0E-05 EPI Acetaldehyde 75-07-0 4.4E+01 PHYSPRO 2.7E-03 6.7E-05 PHYSPROP 9.0E+02 PHYSPROP -1.2E+02 PHYSPROP 7.8E-01 CRC89 1.3E-01 1.4E-05 WATER9 1.0E+00 EPI -3.4E-01 PHYSPRO 1.0E+06 PHYSPROP 1.3E-03 1.9E-01 4.5E-01 5.3E-04 EPI Acetochlor 34256-82-1 2.7E+02 PHYSPRO 9.1E-07 2.2E-08 PHYSPROP 2.8E-05 PHYSPROP 1.1E+01 PubChem 1.1E+00 PubChem 2.2E-02 5.6E-06 WATER9 3.0E+02 EPI 3.0E+00 PHYSPRO 2.2E+02 PHYSPROP 3.1E-02 3.4E+00 8.2E+00 5.0E-03 EPI Acetone 67-64-1 5.8E+01 PHYSPRO 1.4E-03 3.5E-05 PHYSPROP 2.3E+02 PHYSPROP -9.5E+01 PHYSPROP 7.8E-01 CRC89 1.1E-01 1.2E-05 WATER9 2.4E+00 EPI -2.4E-01 PHYSPRO 1.0E+06 PHYSPROP 1.5E-03 2.2E-01 5.3E-01 5.1E-04 EPI Acetone Cyanohydrin 75-86-5 8.5E+01 PHYSPRO 8.1E-08 2.0E-09 PHYSPROP 3.4E-01 PHYSPROP -1.9E+01 PHYSPROP 9.3E-01 CRC89 8.6E-02 1.0E-05 WATER9 1.0E+00 -

Environmental Health Criteria 206 Methyl Tertiary-Butyl Ether -J
INTERNATIONAL PROGRAMME ON CHEMICAL SAFETY Environmental Health Criteria 206 Methyl tertiary-Butyl Ether -J. l \r LNTER-ORGAN1ZATON PROGRAMME FOR THE SOUND MANAGEMENT OF CHEMICALS I JiVJ A cooperative agreement among UNEP, ILO, FAO, WHO, UNIDO, UNITAR and OECD THE ENVIRONMENTAL HEALTH CRITERIA SERIES Acetaldehyde (No. 167, 1995) Chiorofluorocarbons, partially halogenated Acetonitrile (No. 154, 1993) (ethane derivatives) (No. 139, 1992) Acrolein (No. 127, 1991) (methane derivatives) (No. 126, 1991) Acrylamide (No. 49, 1985) Chloroform (No. 163, 1994) Acrylic acid (No. 191, 1997) Chlorophenols (No. 93, 1989) Acrylonitrile (No. 28, 1983) Chlorothalonil (No. 183, 1996) Aged population, principles for evaluating Chromium (No. 61, 1988) the effects of chemicals (No. 144, 1992) Chrysotile asbestos (No. 203, 1998) Aldicarb (No. 121, 1991) Copper (No. 200, 1998) Aldrin and dieldrin (No. 91, 1989) Cresols (No. 168, 1995) Allethrins (No. 87, 1989) Cyhalothrin (No. 99, 1990) Aluminium (No. 194, 1997) Cypermethrin (No. 82, 1989) Amitrole (No. 158, 1994) Cypermethrin, alpha- (No. 142, 1992) Ammonia (No. 54, 1986) DOT and its derivatives (No. 9, 1979) Anticoagulant rodenticides (No. 175, 1995) DOT and its derivatives - Arsenic (No. 18, 1981) environmental aspects (No, 83, 1989) Asbestos and other natural mineral fibres Deltamethrjn (No. 97, 1990) (No. 53, 1986) Demeton-S-methyl (No. 197, 1997) Barium (No. 107, 1990) Diaminotoluenes (No. 74, 1987) Benomyl (No. 148, 1993) Diazinon (No. 198, 1997) Benzene (No. 150, 1993) 1,2-Dibromoethane (No. 177, 1996) Beryllium (No. 106, 1990) Oi-n-butyl phthalate (No. 189, 1997) Biomarkers and risk assessment: concepts 1 ,2-Dichloroethane and principles (No. 155, 1993) (No. 62, 1987, 1St edition) Blotoxins, aquatic (marine and freshwater) (No. -

TOXICOLOGICAL PROFILE for METHYL Tert-BUTYL ETHER
TOXICOLOGICAL PROFILE FOR METHYL tert-BUTYL ETHER U.S. DEPARTMENT OF HEALTH AND HUMAN SERVICES Public Health Service Agency for Toxic Substances and Disease Registry August 1996 METHYL tert-BUTYL ETHER ii DISCLAIMER The use of company or product name(s) is for identification only and does not imply endorsement by the Agency for Toxic Substances and Disease Registry. METHYL tert-BUTYL ETHER iii UPDATE STATEMENT Toxicological profiles are revised and republished as necessary, but no less than once every three years. For information regarding the update status of previously released profiles, contact ATSDR at: Agency for Toxic Substances and Disease Registry Division of Toxicology/Toxicology Information Branch 1600 Clifton Road NE, E-29 Atlanta, Georgia 30333 vi *Legislative Background The toxicological profiles are developed in response to the Superfund Amendments and Reauthorization Act (SARA) of 1986 (Public Law 99-499) which amended the Comprehensive Environmental Response, Compensation, and Liability Act of 1980 (CERCLA or Superfund). This public law directed ATSDR to prepare toxicological profiles for hazardous substances most commonly found at facilities on the CERCLA National Priorities List and that pose the most significant potential threat to human health, as determined by ATSDR and the EPA. The availability of the revised priority list of 275 hazardous substances was announced in the Federal Register on April 29, 1996 (61 FR 18744). For prior versions of the list of substances, see Federal Register notices dated April 17, 1987 (52 FR 12866); October 20, 1988 (53 FR 41280); October 26, 1989 (54 FR 43619); October 17, 1990 (55 FR 42067); October 17, 199l (56 FR 52166); October 28, 1992 (57 FR 48801); and February 28, 1994 (59 FR 9486). -

The Effect of Dosing Regimen on Outcomes of Vitamin D Supplementation Trials a Study of Current Literature
Faculty of Medicine The effect of dosing regimen on outcomes of vitamin D supplementation trials A study of current literature — Anette Uhlving Larsen Master thesis in Medicine (MED-3950) June 2019 Supervisor: Professor Rolf Jorde, Institute of Clinical Medicine Abstract Results from observational studies have indicated associations between vitamin D and extra- skeletal outcomes, including respiratory tract infections (RTI) and all-cause mortality. However, available trial-data have shown inconsistent results. The main objective of this thesis was to investigate whether a beneficial effect of daily supplementation of vitamin D on RTI and all-cause mortality could have been masked by the use of less frequent supplementation intervals. This thesis included data from double-blinded, randomized controlled trials (RCTs) published in the last 10 years. Eligible trials were identified through screening of the reference lists of systematic reviews of meta-analyses (MAs), and of reference lists of MAs on the selected outcomes included in these reviews. Also, additional searches were performed to ensure that also recently published RCTs, not identified in a previous step of the search strategy, were considered for inclusion. The search strategy was designed to promote selection of trials of adequate methodological quality. To be included the record had to be written in English and report results of a double-blinded placebo-controlled RCT with vitamin D supplementation in a human population. Studies including pregnant women or assessing the effect of prenatal supplementation were not included, nor were studies including populations with chronic kidney disease and/or other diseases known to affect the conversion of active metabolites of vitamin D. -

Focus on China TOP ARTICLES SUPPLEMENT
Powered by Focus on China TOP ARTICLES SUPPLEMENT CONTENTS RESEARCH ARTICLE: Simultaneous determination of hyzetimibe and its main active metabolite in plasma by LC–MS/MS and its application in PK study Bioanalysis Vol. 7 Issue 15 RESEARCH ARTICLE: Highly specific and sensitive immunoassay for the measurement of prostaglandin E2 in biological fluids Bioanalysis Vol. 7 Issue 19 REVIEW: Derivatization methods for LC–MS analysis of endogenous compounds Bioanalysis Vol. 7 Issue 19 METHODOLOGY: Development of an Excel-based laboratory information management system for improving workflow efficiencies in early ADME screening Bioanalysis Vol. 8 Issue 2 Research Article For reprint orders, please contact [email protected] 7 Research Article 2015/07/30 Simultaneous determination of hyzetimibe and its main active metabolite in plasma by LC–MS/MS and its application in PK study Bioanalysis Background: Hyzetimibe is a new compound belonging to a novel class of selective Jinliang Chen1, Honggang cholesterol absorption inhibitors. A simple, highly sensitive LC–MS/MS method has Lou1, Bo Jiang1, Rong Shao1, been developed for the quantification of hyzetimibe and its main active metabolite, Zourong Ruan1 ,1 hyzetimibe-glucuronide, in human plasma. Results: Analytical samples were prepared & Jian’an Wang* 1Center of Clinical Pharmacology, using a protein precipitation method coupled with a concentration process. The the Second Affiliated Hospital of linearity of this method was established for concentrations in the ranges of 0.05–50 Zhejiang University School of Medicine, and 0.5–500 ng/ml for hyzetimibe and hyzetimibe-glucuronide, respectively. The Hangzhou, Zhejiang, China accuracy and precision of the method varied from 97.9 to 105% and 2.6 to 7.4%, *Author for correspondence: respectively. -

G Protein-Coupled Estrogen Receptor in Cancer and Stromal Cells: Functions and Novel Therapeutic Perspectives
cells Review G Protein-Coupled Estrogen Receptor in Cancer and Stromal Cells: Functions and Novel Therapeutic Perspectives Richard A. Pepermans 1, Geetanjali Sharma 1,2 and Eric R. Prossnitz 1,2,3,* 1 Division of Molecular Medicine, Department of Internal Medicine, University of New Mexico Health Sciences Center, Albuquerque, NM 87131, USA; [email protected] (R.A.P.); [email protected] (G.S.) 2 Center of Biomedical Research Excellence in Autophagy, Inflammation and Metabolism, University of New Mexico Health Sciences Center, Albuquerque, NM 87131, USA 3 University of New Mexico Comprehensive Cancer Center, University of New Mexico Health Sciences Center, Albuquerque, NM 87131, USA * Correspondence: [email protected]; Tel.: +1-505-272-5647 Abstract: Estrogen is involved in numerous physiological and pathophysiological systems. Its role in driving estrogen receptor-expressing breast cancers is well established, but it also has important roles in a number of other cancers, acting both on tumor cells directly as well as in the function of multiple cells of the tumor microenvironment, including fibroblasts, immune cells, and adipocytes, which can greatly impact carcinogenesis. One of its receptors, the G protein-coupled estrogen receptor (GPER), has gained much interest over the last decade in both health and disease. Increasing evidence shows that GPER contributes to clinically observed endocrine therapy resistance in breast cancer while also playing a complex role in a number of other cancers. Recent discoveries regarding the targeting of GPER in combination with immune checkpoint inhibition, particularly in melanoma, have led to the initiation of the first Phase I clinical trial for the GPER-selective agonist G-1. -

2019 Complete Drug List (Formulary)
20191 COMPLETE DRUG LIST (FORMULARY) Prescription drug list information UnitedHealthcare® Group Medicare Advantage Important Notes: This document has information about the drugs covered by this plan. For more up-to-date information or if you have any questions, please call Customer Service at: Toll-free 1-800-457-8506, TTY 711 8 a.m. - 8 p.m. local time, Monday - Friday www.UHCRetiree.com Formulary ID Number 00019090, Version 9 Y0066_180629_042324 Last updated October 1, 2018 TABLE2 11 OF CONTENTS What is a drug list?......................................................................................................................................3 Note to existing members.......................................................................................................................... 3 How do I use the drug list?.........................................................................................................................4 What are generic drugs?............................................................................................................................ 4 Drug payment stage and drug tiers...........................................................................................................5 Getting Extra Help.......................................................................................................................................5 Are there any rules or limits on my drug coverage?................................................................................ 6 What if my drug is not -

Diethylstilbestrol Activates Catsper and Disturbs Progesterone Actions in Human Spermatozoa
Human Reproduction, Vol.32, No.2 pp. 290–298, 2017 Advanced Access publication on December 28, 2016 doi:10.1093/humrep/dew332 ORIGINAL ARTICLE Andrology Diethylstilbestrol activates CatSper and disturbs progesterone actions in human spermatozoa † † Qian-xing Zou1, , Zhen Peng1, , Qing Zhao1, Hou-yang Chen2, Yi-min Cheng1, Qing Liu1, Yuan-qiao He1, Shi-qi Weng1, Hua-feng Wang1, Tao Wang1, Li-ping Zheng1, and Tao Luo1,* 1Institute of Life Science and School of Life Science, Nanchang University, Nanchang, Jiangxi 330031, PR China 2Reproductive Medical Center, Jiangxi Provincial Maternal and Child Health Hospital, Nanchang, Jiangxi 330006, PR China *Correspondence address. Institute of Life Science and School of Life Science, Nanchang University, Nanchang, Jiangxi 330031, PR China. E-mail: [email protected] Submitted on June 24, 2016; resubmitted on November 24, 2016; accepted on December 16, 2016 STUDY QUESTION: Is diethylstilbestrol (DES), a prototypical endocrine-disrupting chemical (EDC), able to induce physiological changes in human spermatozoa and affect progesterone actions? + SUMMARY ANSWER: DES promoted Ca2 flux into human spermatozoa by activating the cation channel of sperm (CatSper) and sup- + pressed progesterone-induced Ca2 signaling, tyrosine phosphorylation and sperm functions. WHAT IS KNOWN ALREADY: DES significantly impairs the male reproductive system both in fetal and postnatal exposure. Although various EDCs affect human spermatozoa in a non-genomic manner, the effect of DES on human spermatozoa remains unknown. STUDY DESIGN, SIZE, DURATION: Sperm samples from normozoospermic donors were exposed in vitro to a range of DES concen- trations with or without progesterone at 37°C in a 5% CO2 incubator to mimic the putative exposure to this toxicant in seminal plasma and the female reproductive tract fluids. -

Clinical Practice Guidelines for Managing Dyslipidemias in Kidney
American Journal of Transplantation 2004; 4 (Suppl. 7): 13–53 Copyright # Blackwell Munksgaard 2004 Blackwell Munksgaard Clinical practice guidelines for managing dyslipidemias in kidney transplant patients: a report from the Managing Dyslipidemias in Chronic Kidney Disease Work Group of the National Kidney Foundation Kidney Disease Outcomes Quality Initiative National Kidney Foundation kidney transplant patients. These guidelines are divided WorkGroupMembers:B.Kasiske,F.G.Cosio, into four sections. The first section (Introduction) pro- J.Beto,K.Bolton,B.M.Chavers,R.GrimmJr., vides the rationale for the guidelines, and describes the target population, scope, intended users, and methods. A. Levin, B. Masri, R. Parekh, C. Wanner, The second section presents guidelines on the assess- D.C. Wheeler and P.W.F. Wilson ment of dyslipidemias (guidelines 1–3), while the third section offers guidelines for the treatment of dyslipid- Correspondence: Karen Glowacki, National Kidney emias (guidelines 4–5). The key guideline statements Foundation 30 E. 33rd Street, 11th Fl. New York, are supported mainly by data from studies in the general NY 10016, USA; email: [email protected] population, but there is an urgent need for additional studies in CKD and in transplant patients. Therefore, the last section outlines recommendations for research. ABSTRACT The incidence of cardiovascular disease (CVD) is very high in patients with chronic kidney (CKD) disease and in kidney transplant recipients. Indeed, available evi- INTRODUCTION dence for these patients suggests that the 10-year cumulative risk of coronary heart disease is at least The rationale for these guidelines 20%, or roughly equivalent to the risk seen in patients These guidelines are the result of an ongoing effort by the with previous CVD. -

Vitamin D Supplementation for Prevention of Mortality in Adults (Review)
Cochrane Database of Systematic Reviews Vitamin D supplementation for prevention of mortality in adults (Review) Bjelakovic G, Gluud LL, Nikolova D, Whitfield K, Wetterslev J, Simonetti RG, Bjelakovic M, Gluud C Bjelakovic G, Gluud LL, Nikolova D, Whitfield K, Wetterslev J, Simonetti RG, Bjelakovic M, Gluud C. Vitamin D supplementation for prevention of mortality in adults. Cochrane Database of Systematic Reviews 2014, Issue 1. Art. No.: CD007470. DOI: 10.1002/14651858.CD007470.pub3. www.cochranelibrary.com Vitamin D supplementation for prevention of mortality in adults (Review) Copyright © 2014 The Cochrane Collaboration. Published by John Wiley & Sons, Ltd. TABLE OF CONTENTS HEADER....................................... 1 ABSTRACT ...................................... 1 PLAINLANGUAGESUMMARY . 2 SUMMARY OF FINDINGS FOR THE MAIN COMPARISON . ..... 4 BACKGROUND .................................... 8 OBJECTIVES ..................................... 9 METHODS ...................................... 9 RESULTS....................................... 13 Figure1. ..................................... 14 Figure2. ..................................... 17 Figure3. ..................................... 18 Figure4. ..................................... 21 Figure5. ..................................... 22 Figure6. ..................................... 24 DISCUSSION ..................................... 26 AUTHORS’CONCLUSIONS . 31 ACKNOWLEDGEMENTS . 31 REFERENCES ..................................... 31 CHARACTERISTICSOFSTUDIES . 56 DATAANDANALYSES.