A Calixpyrrole Derivative Acts As an Antagonist to GPER, a G-Protein
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Cyproterone Art. 31
Приложение I Списък на лекарствените продукти и форми 1 Държава членка Притежател на Наименование на INN/Активно Фармацевтична Начин на (ЕИП) разрешението за продукта вещество + форма приложение употреба Количество на активното вещество (в дозова единица) Австрия Bayer Austria Gmbh Androcur Depot Cyproterone Acetate Инжекционен Интрамускулно 300mg/3ml разтвор приложение Австрия Bayer Austria Gmbh Climen Cyproterone Acetate 1mg Обвита таблетка Перорално таблетка, Estradiol приложение Valerate 2mg таблетка| Estradiol Valerate 2mg таблетка Австрия Bayer Austria Gmbh Climen 28-Tage Cyproterone Acetate 1mg Обвита таблетка Перорално таблетка, Estradiol приложение Valerate 2mg таблетка| Estradiol Valerate 2mg таблетка Австрия Bayer Austria Gmbh Diane Mite Cyproterone Acetate 2mg Обвита таблетка Перорално таблетка, Ethinylestradiol приложение 35μg таблетка Австрия Bayer Austria Gmbh Minerva Cyproterone Acetate 2mg Обвита таблетка Перорално таблетка, Ethinylestradiol приложение 0,035mg таблетка Австрия Bayer Austria Gmbh Andro-Diane Cyproterone Acetate Таблетка Перорално 10mg таблетка приложение Австрия Bayer Austria Gmbh Androcur Cyproterone Acetate Таблетка Перорално 100mg таблетка приложение Австрия Bayer Austria Gmbh Androcur Cyproterone Acetate Таблетка Перорално 50mg таблетка приложение Австрия Gynial Gmbh Alisma Cyproterone Acetate 2mg Филмирана таблетка Перорално таблетка, Ethinylestradiol приложение 35μg таблетка 2 Държава членка Притежател на Наименование на INN/Активно Фармацевтична Начин на (ЕИП) разрешението за продукта вещество + -

Aldosterone Enhances Renal Calcium Reabsorption by Two Types of Channels
View metadata, citation and similar papers at core.ac.uk brought to you by CORE provided by Elsevier - Publisher Connector Kidney International, Vol. 66 (2004), pp. 242–250 Aldosterone enhances renal calcium reabsorption by two types of channels MARIE LECLERC,MICHELE` G. BRUNETTE, and DENIS COUCHOUREL Maisonneuve-Rosemont Hospital and University of Montreal, Canada Aldosterone enhances renal calcium reabsorption by two types transepithelial potential difference in the late segments of channels. of the nephron, reflecting a stimulation of the amiloride- Background. Aldosterone has been known for many years + + sensitive Na transport by the apical membranes. to increase sodium (Na ) reabsorption by the distal nephron. The present in vitro experiments investigated the effect of the In contrast to the abundant literature dealing with the + hormone on calcium (Ca2 ) transport by the luminal membrane antinatriuretic effect of aldosterone, data concerning its + of the rabbit nephron, independent of any systemic influence. action on Ca2 transport are relatively scarce. Clearance Methods. Proximal and distal tubules were incubated with studies showed either an increase in calciuria [2] or an + either aldosterone or the carrier. The luminal membranes of absence of significant change of Ca2 excretion [3, 4] af- these tubules were purified, vesiculated, and 45Ca uptake by these vesicles was subsequently measured. ter aldosterone administration. In a randomized study in Results. Treatment of the distal tubules with 10−8 mol/L al- male volunteers, Van Hamersvelt et al [5] compared the + + dosterone enhanced both 0.1 and 0.5 mmol/L Ca2 transport. effect of the Ca2 channel blocker felodipine, alone or The hormone action was abolished by tyrosine kinase inhibitors. -

Targeting Lysophosphatidic Acid in Cancer: the Issues in Moving from Bench to Bedside
View metadata, citation and similar papers at core.ac.uk brought to you by CORE provided by IUPUIScholarWorks cancers Review Targeting Lysophosphatidic Acid in Cancer: The Issues in Moving from Bench to Bedside Yan Xu Department of Obstetrics and Gynecology, Indiana University School of Medicine, 950 W. Walnut Street R2-E380, Indianapolis, IN 46202, USA; [email protected]; Tel.: +1-317-274-3972 Received: 28 August 2019; Accepted: 8 October 2019; Published: 10 October 2019 Abstract: Since the clear demonstration of lysophosphatidic acid (LPA)’s pathological roles in cancer in the mid-1990s, more than 1000 papers relating LPA to various types of cancer were published. Through these studies, LPA was established as a target for cancer. Although LPA-related inhibitors entered clinical trials for fibrosis, the concept of targeting LPA is yet to be moved to clinical cancer treatment. The major challenges that we are facing in moving LPA application from bench to bedside include the intrinsic and complicated metabolic, functional, and signaling properties of LPA, as well as technical issues, which are discussed in this review. Potential strategies and perspectives to improve the translational progress are suggested. Despite these challenges, we are optimistic that LPA blockage, particularly in combination with other agents, is on the horizon to be incorporated into clinical applications. Keywords: Autotaxin (ATX); ovarian cancer (OC); cancer stem cell (CSC); electrospray ionization tandem mass spectrometry (ESI-MS/MS); G-protein coupled receptor (GPCR); lipid phosphate phosphatase enzymes (LPPs); lysophosphatidic acid (LPA); phospholipase A2 enzymes (PLA2s); nuclear receptor peroxisome proliferator-activated receptor (PPAR); sphingosine-1 phosphate (S1P) 1. -
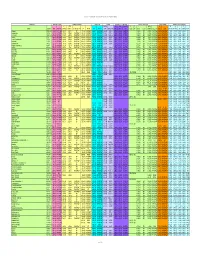
Chemical-Specific Parameters Supporting Table May 2016 Analyte
Regional Screening Level (RSL) Chemical-specific Parameters Supporting Table May 2016 Contaminant Molecular Weight Volatility Parameters Melting Point Density Diffusivity in Air and Water Partition Coefficients Water Solubility Tapwater Dermal Parameters H` (atm- Density Dia Diw Dia and Diw Kd Kd Koc log Kow S B τevent t* Kp 3 3 2 2 Analyte CAS No. MW MW Ref (unitless) m /mole) H` and HLC Ref VP VP Ref MP MP Ref (g/cm ) Density Ref (cm /s) (cm /s) Ref (L/kg) Ref (L/kg) Koc Ref (unitless) log Kow Ref (mg/L) S Ref (unitless) (hr/event) (hr) (cm/hr) K Ref Acephate 30560-19-1 1.8E+02 PHYSPRO 2.0E-11 5.0E-13 EPI 1.7E-06 PHYSPROP 8.8E+01 PHYSPROP 1.4E+00 CRC89 3.7E-02 8.0E-06 WATER9 1.0E+01 EPI -8.5E-01 PHYSPRO 8.2E+05 PHYSPROP 2.1E-04 1.1E+00 2.7E+00 4.0E-05 EPI Acetaldehyde 75-07-0 4.4E+01 PHYSPRO 2.7E-03 6.7E-05 PHYSPROP 9.0E+02 PHYSPROP -1.2E+02 PHYSPROP 7.8E-01 CRC89 1.3E-01 1.4E-05 WATER9 1.0E+00 EPI -3.4E-01 PHYSPRO 1.0E+06 PHYSPROP 1.3E-03 1.9E-01 4.5E-01 5.3E-04 EPI Acetochlor 34256-82-1 2.7E+02 PHYSPRO 9.1E-07 2.2E-08 PHYSPROP 2.8E-05 PHYSPROP 1.1E+01 PubChem 1.1E+00 PubChem 2.2E-02 5.6E-06 WATER9 3.0E+02 EPI 3.0E+00 PHYSPRO 2.2E+02 PHYSPROP 3.1E-02 3.4E+00 8.2E+00 5.0E-03 EPI Acetone 67-64-1 5.8E+01 PHYSPRO 1.4E-03 3.5E-05 PHYSPROP 2.3E+02 PHYSPROP -9.5E+01 PHYSPROP 7.8E-01 CRC89 1.1E-01 1.2E-05 WATER9 2.4E+00 EPI -2.4E-01 PHYSPRO 1.0E+06 PHYSPROP 1.5E-03 2.2E-01 5.3E-01 5.1E-04 EPI Acetone Cyanohydrin 75-86-5 8.5E+01 PHYSPRO 8.1E-08 2.0E-09 PHYSPROP 3.4E-01 PHYSPROP -1.9E+01 PHYSPROP 9.3E-01 CRC89 8.6E-02 1.0E-05 WATER9 1.0E+00 -

Glucocorticoid Regulation of the G-Protein Coupled Estrogen Receptor (GPER) in Mouse Hippocampal Neurons
Glucocorticoid Regulation of the G-Protein Coupled Estrogen Receptor (GPER) in Mouse Hippocampal Neurons by Kate Colleen Eliza Nicholson A Thesis presented to The University of Guelph In partial fulfilment of requirements for the degree of Master of Science in Biomedical Sciences Guelph, Ontario, Canada © Kate Colleen Eliza Nicholson, August, 2019 ABSTRACT GLUCOCORTICOID REGULATION OF THE G-PROTEIN COUPLED ESTROGEN RECEPTOR (GPER) IN MOUSE HIPPOCAMPAL NEURONS Kate Colleen Eliza Nicholson Advisor: University of Guelph, 2019 Dr. Neil J. MacLusky The most prevalent estrogen, 17β-estradiol, binds the non-classical G-protein coupled estrogen receptor (GPER) with high affinity resulting in rapid activation of the c- jun N terminal kinase (JNK) pathway. GPER activation mediates some of the rapid neurotrophic and memory-enhancing effects of 17β-estradiol in the female hippocampus. However, exposure to stressful stimuli may impair these beneficial effects. This thesis characterizes neurosteroid receptor expression in murine-derived mHippoE cell lines that are subsequently used to investigate the glucocorticoid regulation of GPER protein expression and functional activation. This thesis demonstrates that 24-hour treatment with a glucocorticoid receptor agonist reduces GPER protein expression and activation of JNK in female-derived mHippoE-14s. Using an in vivo model, treatment with glucocorticoids significantly reduces hippocampal activation of JNK in female ovariectomized CD1 mice. Collectively, this thesis uses in vitro and in vivo models to characterize glucocorticoid regulation of GPER expression and signalling in female murine hippocampal neurons. ACKNOWLEDGEMENTS To Dr. MacLusky: I would like to thank you for inspiring my passion for science and pursuit of knowledge. Over the past 2 years, you have provided me with countless opportunities to grow as a young researcher and I am tremendously grateful for this. -

G Protein-Coupled Estrogen Receptor Inhibits the P2Y Receptor-Mediated Ca2+ Signaling Pathway in Human Airway Epithelia
Pflugers Arch - Eur J Physiol DOI 10.1007/s00424-016-1840-7 SIGNALING AND CELL PHYSIOLOGY G protein-coupled estrogen receptor inhibits the P2Y receptor-mediated Ca2+ signaling pathway in human airway epithelia Yuan Hao1 & Alison W. Chow1 & Wallace C. Yip 1 & Chi H. Li1 & Tai F. Wan 1 & Benjamin C. Tong2 & King H. Cheung2 & Wood Y. Chan 1 & Yangchao Chen 1 & Christopher H. Cheng1 & Wing H. Ko1 Received: 21 January 2016 /Revised: 11 May 2016 /Accepted: 22 May 2016 # The Author(s) 2016. This article is published with open access at Springerlink.com Abstract P2Y receptor activation causes the release of in- inhibitory effects of G1 or E2 on P2Y receptor-mediated flammatory cytokines in the bronchial epithelium, whereas Ca2+ mobilization and cytokine secretion were due to G protein-coupled estrogen receptor (GPER), a novel estrogen GPER-mediated activation of a cAMP-dependent PKA path- (E2) receptor, may play an anti-inflammatory role in this pro- way. This study has reported, for the first time, the expression cess. We investigated the cellular mechanisms underlying the and function of GPER as an anti-inflammatory component in inhibitory effect of GPER activation on the P2Y receptor- human bronchial epithelia, which may mediate through its mediated Ca2+ signaling pathway and cytokine production in opposing effects on the pro‐inflammatory pathway activated airway epithelia. Expression of GPER in primary human by the P2Y receptors in inflamed airway epithelia. bronchial epithelial (HBE) or 16HBE14o- cells was con- firmed on both the mRNA and protein levels. Stimulation of Keywords GPER . P2Y receptor signaling pathway . Human HBE or 16HBE14o- cells with E2 or G1, a specific agonist of . -

Environmental Health Criteria 206 Methyl Tertiary-Butyl Ether -J
INTERNATIONAL PROGRAMME ON CHEMICAL SAFETY Environmental Health Criteria 206 Methyl tertiary-Butyl Ether -J. l \r LNTER-ORGAN1ZATON PROGRAMME FOR THE SOUND MANAGEMENT OF CHEMICALS I JiVJ A cooperative agreement among UNEP, ILO, FAO, WHO, UNIDO, UNITAR and OECD THE ENVIRONMENTAL HEALTH CRITERIA SERIES Acetaldehyde (No. 167, 1995) Chiorofluorocarbons, partially halogenated Acetonitrile (No. 154, 1993) (ethane derivatives) (No. 139, 1992) Acrolein (No. 127, 1991) (methane derivatives) (No. 126, 1991) Acrylamide (No. 49, 1985) Chloroform (No. 163, 1994) Acrylic acid (No. 191, 1997) Chlorophenols (No. 93, 1989) Acrylonitrile (No. 28, 1983) Chlorothalonil (No. 183, 1996) Aged population, principles for evaluating Chromium (No. 61, 1988) the effects of chemicals (No. 144, 1992) Chrysotile asbestos (No. 203, 1998) Aldicarb (No. 121, 1991) Copper (No. 200, 1998) Aldrin and dieldrin (No. 91, 1989) Cresols (No. 168, 1995) Allethrins (No. 87, 1989) Cyhalothrin (No. 99, 1990) Aluminium (No. 194, 1997) Cypermethrin (No. 82, 1989) Amitrole (No. 158, 1994) Cypermethrin, alpha- (No. 142, 1992) Ammonia (No. 54, 1986) DOT and its derivatives (No. 9, 1979) Anticoagulant rodenticides (No. 175, 1995) DOT and its derivatives - Arsenic (No. 18, 1981) environmental aspects (No, 83, 1989) Asbestos and other natural mineral fibres Deltamethrjn (No. 97, 1990) (No. 53, 1986) Demeton-S-methyl (No. 197, 1997) Barium (No. 107, 1990) Diaminotoluenes (No. 74, 1987) Benomyl (No. 148, 1993) Diazinon (No. 198, 1997) Benzene (No. 150, 1993) 1,2-Dibromoethane (No. 177, 1996) Beryllium (No. 106, 1990) Oi-n-butyl phthalate (No. 189, 1997) Biomarkers and risk assessment: concepts 1 ,2-Dichloroethane and principles (No. 155, 1993) (No. 62, 1987, 1St edition) Blotoxins, aquatic (marine and freshwater) (No. -

G Protein-Coupled Receptors Function As Cell Membrane Receptors for the Steroid Hormone 20-Hydroxyecdysone Xiao-Fan Zhao
Zhao Cell Communication and Signaling (2020) 18:146 https://doi.org/10.1186/s12964-020-00620-y REVIEW Open Access G protein-coupled receptors function as cell membrane receptors for the steroid hormone 20-hydroxyecdysone Xiao-Fan Zhao Abstract G protein-coupled receptors (GPCRs) are cell membrane receptors for various ligands. Recent studies have suggested that GPCRs transmit animal steroid hormone signals. Certain GPCRs have been shown to bind steroid hormones, for example, G protein-coupled estrogen receptor 1 (GPER1) binds estrogen in humans, and Drosophila dopamine/ecdysteroid receptor (DopEcR) binds the molting hormone 20-hydroxyecdysone (20E) in insects. This review summarizes the research progress on GPCRs as animal steroid hormone cell membrane receptors, including the nuclear and cell membrane receptors of steroid hormones in mammals and insects, the 20E signaling cascade via GPCRs, termination of 20E signaling, and the relationship between genomic action and the nongenomic action of 20E. Studies indicate that 20E induces a signal via GPCRs to regulate rapid cellular responses, including rapid Ca2+ release from the endoplasmic reticulum and influx from the extracellular medium, as well as rapid protein phosphorylation and subcellular translocation. 20E via the GPCR/Ca2+/PKC/signaling axis and the GPCR/cAMP/PKA- signaling axis regulates gene transcription by adjusting transcription complex formation and DNA binding activity. GPCRs can bind 20E in the cell membrane and after being isolated, suggesting GPCRs as cell membrane receptors of 20E. This review deepens our understanding of GPCRs as steroid hormone cell membrane receptors and the GPCR-mediated signaling pathway of 20E (20E-GPCR pathway), which will promote further study of steroid hormone signaling via GPCRs, and presents GPCRs as targets to explore new pharmaceutical materials to treat steroid hormone-related diseases or control pest insects. -

Download (PDF)
Supplemental Information Biological and Pharmaceutical Bulletin Promoter Methylation Profiles between Human Lung Adenocarcinoma Multidrug Resistant A549/Cisplatin (A549/DDP) Cells and Its Progenitor A549 Cells Ruiling Guo, Guoming Wu, Haidong Li, Pin Qian, Juan Han, Feng Pan, Wenbi Li, Jin Li, and Fuyun Ji © 2013 The Pharmaceutical Society of Japan Table S1. Gene categories involved in biological functions with hypomethylated promoter identified by MeDIP-ChIP analysis in lung adenocarcinoma MDR A549/DDP cells compared with its progenitor A549 cells Different biological Genes functions transcription factor MYOD1, CDX2, HMX1, THRB, ARNT2, ZNF639, HOXD13, RORA, FOXO3, HOXD10, CITED1, GATA1, activity HOXC6, ZGPAT, HOXC8, ATOH1, FLI1, GATA5, HOXC4, HOXC5, PHTF1, RARB, MYST2, RARG, SIX3, FOXN1, ZHX3, HMG20A, SIX4, NR0B1, SIX6, TRERF1, DDIT3, ASCL1, MSX1, HIF1A, BAZ1B, MLLT10, FOXG1, DPRX, SHOX, ST18, CCRN4L, TFE3, ZNF131, SOX5, TFEB, MYEF2, VENTX, MYBL2, SOX8, ARNT, VDR, DBX2, FOXQ1, MEIS3, HOXA6, LHX2, NKX2-1, TFDP3, LHX6, EWSR1, KLF5, SMAD7, MAFB, SMAD5, NEUROG1, NR4A1, NEUROG3, GSC2, EN2, ESX1, SMAD1, KLF15, ZSCAN1, VAV1, GAS7, USF2, MSL3, SHOX2, DLX2, ZNF215, HOXB2, LASS3, HOXB5, ETS2, LASS2, DLX5, TCF12, BACH2, ZNF18, TBX21, E2F8, PRRX1, ZNF154, CTCF, PAX3, PRRX2, CBFA2T2, FEV, FOS, BARX1, PCGF2, SOX15, NFIL3, RBPJL, FOSL1, ALX1, EGR3, SOX14, FOXJ1, ZNF92, OTX1, ESR1, ZNF142, FOSB, MIXL1, PURA, ZFP37, ZBTB25, ZNF135, HOXC13, KCNH8, ZNF483, IRX4, ZNF367, NFIX, NFYB, ZBTB16, TCF7L1, HIC1, TSC22D1, TSC22D2, REXO4, POU3F2, MYOG, NFATC2, ENO1, -

Sex Steroids Regulate Skin Pigmentation Through Nonclassical
RESEARCH ARTICLE Sex steroids regulate skin pigmentation through nonclassical membrane-bound receptors Christopher A Natale1, Elizabeth K Duperret1, Junqian Zhang1, Rochelle Sadeghi1, Ankit Dahal1, Kevin Tyler O’Brien2, Rosa Cookson2, Jeffrey D Winkler2, Todd W Ridky1* 1Department of Dermatology, Perelman School of Medicine, University of Pennsylvania, Philadelphia, United States; 2Department of Chemistry, University of Pennsylvania, Philadelphia, United States Abstract The association between pregnancy and altered cutaneous pigmentation has been documented for over two millennia, suggesting that sex hormones play a role in regulating epidermal melanocyte (MC) homeostasis. Here we show that physiologic estrogen (17b-estradiol) and progesterone reciprocally regulate melanin synthesis. This is intriguing given that we also show that normal primary human MCs lack classical estrogen or progesterone receptors (ER or PR). Utilizing both genetic and pharmacologic approaches, we establish that sex steroid effects on human pigment synthesis are mediated by the membrane-bound, steroid hormone receptors G protein-coupled estrogen receptor (GPER), and progestin and adipoQ receptor 7 (PAQR7). Activity of these receptors was activated or inhibited by synthetic estrogen or progesterone analogs that do not bind to ER or PR. As safe and effective treatment options for skin pigmentation disorders are limited, these specific GPER and PAQR7 ligands may represent a novel class of therapeutics. DOI: 10.7554/eLife.15104.001 *For correspondence: ridky@mail. -

TOXICOLOGICAL PROFILE for METHYL Tert-BUTYL ETHER
TOXICOLOGICAL PROFILE FOR METHYL tert-BUTYL ETHER U.S. DEPARTMENT OF HEALTH AND HUMAN SERVICES Public Health Service Agency for Toxic Substances and Disease Registry August 1996 METHYL tert-BUTYL ETHER ii DISCLAIMER The use of company or product name(s) is for identification only and does not imply endorsement by the Agency for Toxic Substances and Disease Registry. METHYL tert-BUTYL ETHER iii UPDATE STATEMENT Toxicological profiles are revised and republished as necessary, but no less than once every three years. For information regarding the update status of previously released profiles, contact ATSDR at: Agency for Toxic Substances and Disease Registry Division of Toxicology/Toxicology Information Branch 1600 Clifton Road NE, E-29 Atlanta, Georgia 30333 vi *Legislative Background The toxicological profiles are developed in response to the Superfund Amendments and Reauthorization Act (SARA) of 1986 (Public Law 99-499) which amended the Comprehensive Environmental Response, Compensation, and Liability Act of 1980 (CERCLA or Superfund). This public law directed ATSDR to prepare toxicological profiles for hazardous substances most commonly found at facilities on the CERCLA National Priorities List and that pose the most significant potential threat to human health, as determined by ATSDR and the EPA. The availability of the revised priority list of 275 hazardous substances was announced in the Federal Register on April 29, 1996 (61 FR 18744). For prior versions of the list of substances, see Federal Register notices dated April 17, 1987 (52 FR 12866); October 20, 1988 (53 FR 41280); October 26, 1989 (54 FR 43619); October 17, 1990 (55 FR 42067); October 17, 199l (56 FR 52166); October 28, 1992 (57 FR 48801); and February 28, 1994 (59 FR 9486). -

I GPER/GPR30 ESTROGEN RECEPTOR
GPER/GPR30 ESTROGEN RECEPTOR: A TARGET FOR PAIN MODULATION A Dissertation Submitted to The Temple University Graduate Board In Partial Fulfillment of the Requirements for the Degree of Doctor of Philosophy By Elena Deliu May, 2012 Examination Committee Members: Dr. Nae J Dun (Advisor), Dept. of Pharmacology, Temple University Dr. Mary E Abood, Dept. of Anatomy and Cell Biology, Temple University Dr. Barrie Ashby, Dept. of Pharmacology, Temple University Dr. Eugen Brailoiu, Dept. of Pharmacology, Temple University Dr. Ellen M Unterwald, Dept. of Pharmacology, Temple University Dr. Hreday Sapru, (External Examiner), Depts. of Neurosciences, Neurosurgery & Pharmacology/Physiology, UMDNJ-NJMS i © 2012 By Elena Deliu All Rights Reserved ii ABSTRACT GPER/GPR30 ESTROGEN RECEPTOR: A TARGET FOR PAIN MODULATION Elena Deliu Doctor of Philosophy Temple University, 2012 Doctoral Advisory Committee Chair: Nae J. Dun, Ph.D. The G protein-coupled estrogen receptor GPER/GPER1, also known as GPR30, was originally cloned as an orphan receptor and later shown to be specifically activated by 17- β-estradiol. This has led to its classification as an estrogen receptor and expanded the perspective on the mechanisms underlying the rapid estrogenic effects reported over the years. GPER is strongly expressed in the central nervous system and peripheral tissues and appears to be involved in a wide variety of physiological and pathological processes. Estrogens are known to alter the processing of nociceptive sensory information and analgesic responses in the central nervous system. Both analgesic and pro-nociceptive effects of estrogens have been reported. Some pro-algesic estrogenic responses have a short latency, suggesting a non-genomic mechanism of action.