Ep 1778618 B1
Total Page:16
File Type:pdf, Size:1020Kb

Load more
Recommended publications
-

Gasket Chemical Services Guide
Gasket Chemical Services Guide Revision: GSG-100 6490 Rev.(AA) • The information contained herein is general in nature and recommendations are valid only for Victaulic compounds. • Gasket compatibility is dependent upon a number of factors. Suitability for a particular application must be determined by a competent individual familiar with system-specific conditions. • Victaulic offers no warranties, expressed or implied, of a product in any application. Contact your Victaulic sales representative to ensure the best gasket is selected for a particular service. Failure to follow these instructions could cause system failure, resulting in serious personal injury and property damage. Rating Code Key 1 Most Applications 2 Limited Applications 3 Restricted Applications (Nitrile) (EPDM) Grade E (Silicone) GRADE L GRADE T GRADE A GRADE V GRADE O GRADE M (Neoprene) GRADE M2 --- Insufficient Data (White Nitrile) GRADE CHP-2 (Epichlorohydrin) (Fluoroelastomer) (Fluoroelastomer) (Halogenated Butyl) (Hydrogenated Nitrile) Chemical GRADE ST / H Abietic Acid --- --- --- --- --- --- --- --- --- --- Acetaldehyde 2 3 3 3 3 --- --- 2 --- 3 Acetamide 1 1 1 1 2 --- --- 2 --- 3 Acetanilide 1 3 3 3 1 --- --- 2 --- 3 Acetic Acid, 30% 1 2 2 2 1 --- 2 1 2 3 Acetic Acid, 5% 1 2 2 2 1 --- 2 1 1 3 Acetic Acid, Glacial 1 3 3 3 3 --- 3 2 3 3 Acetic Acid, Hot, High Pressure 3 3 3 3 3 --- 3 3 3 3 Acetic Anhydride 2 3 3 3 2 --- 3 3 --- 3 Acetoacetic Acid 1 3 3 3 1 --- --- 2 --- 3 Acetone 1 3 3 3 3 --- 3 3 3 3 Acetone Cyanohydrin 1 3 3 3 1 --- --- 2 --- 3 Acetonitrile 1 3 3 3 1 --- --- --- --- 3 Acetophenetidine 3 2 2 2 3 --- --- --- --- 1 Acetophenone 1 3 3 3 3 --- 3 3 --- 3 Acetotoluidide 3 2 2 2 3 --- --- --- --- 1 Acetyl Acetone 1 3 3 3 3 --- 3 3 --- 3 The data and recommendations presented are based upon the best information available resulting from a combination of Victaulic's field experience, laboratory testing and recommendations supplied by prime producers of basic copolymer materials. -

Food and Drugs
21 Part 170 to 199 Revised as of April 1, 2001 Food and Drugs Containing a codification of documents of general applicability and future effect As of April 1, 2001 With Ancillaries Published by Office of the Federal Register National Archives and Records Administration A Special Edition of the Federal Register VerDate 11<MAY>2000 11:38 Apr 16, 2001 Jkt 194064 PO 00000 Frm 00001 Fmt 8091 Sfmt 8091 Y:\SGML\194064F.XXX pfrm02 PsN: 194064F U.S. GOVERNMENT PRINTING OFFICE WASHINGTON : 2001 For sale by the Superintendent of Documents, U.S. Government Printing Office Internet: bookstore.gpo.gov Phone: (202) 512-1800 Tax: (202) 512-2250 Mail Stop: SSOP, Washington, DC 20402–0001 VerDate 11<MAY>2000 11:38 Apr 16, 2001 Jkt 194064 PO 00000 Frm 00002 Fmt 8092 Sfmt 8092 Y:\SGML\194064F.XXX pfrm02 PsN: 194064F Table of Contents Page Explanation ................................................................................................ v Title 21: Chapter I—Food and Drug Administration, Department of Health and Human Services (Continued) ................................................. 3 Finding Aids: Material Approved for Incorporation by Reference ............................ 573 Table of CFR Titles and Chapters ....................................................... 587 Alphabetical List of Agencies Appearing in the CFR ......................... 605 Redesignation Table ............................................................................ 615 List of CFR Sections Affected ............................................................. 617 -

Electronic Supplementary Information For
Electronic Supplementary Material (ESI) for Molecular Systems Design & Engineering. This journal is © The Royal Society of Chemistry 2018 Electronic supplementary information for Understanding structural adaptability: a reactant informatics approach to experiment design Rosalind J. Xu, Jacob H. Olshansky, Philip D. F. Adler, Yongjia Huang, Matthew D. Smith, Matthias Zeller,† Joshua Schrier and Alexander J. Norquist Department of Chemistry, Haverford College, Haverford PA 19041 † Department of Chemistry, Purdue University, West Lafayette IN Author to whom correspondence should be addressed. Haverford College 370 Lancaster Avenue Haverford PA, 19041 USA tel (610) 896 2949 fax (610) 896 4963 email [email protected] http://www.haverford.edu/chem/Norquist/ Figure S1. Three dimensional packing of [C4H14N2]2[VO(SeO3)(HSeO3)]6·5H2O (1). Green octahedra represent [VO6] while purple, red, blue, white and gray spheres represent selenium oxygen, nitrogen, carbon and hydrogen atoms, respectively. Selected hydrogen atoms have been removed for clarity. Figure S2. Three dimensional packing of [C8H26N4][VO(SeO3)(HSeO3)]6·6H2O (3). Green octahedra represent [VO6] while purple, red, blue, white and gray spheres represent selenium oxygen, nitrogen, carbon and hydrogen atoms, respectively. Selected hydrogen atoms have been removed for clarity. Figure S3. (a) One-dimensional chain SBU and (b) and (c) two slice connectivities in [C4H12N2][(VO)3(SeO3)(HSeO3)4]·H2O (4). Green polyhedra represent [VO6] and [VO5] while purple, red, and gray spheres represent selenium oxygen, and hydrogen atoms, respectively. Figure S4. 10-fold cross validation results for the decision trees in this study. Figure S5. Decision tree containing the 75 historical reactions and the 128 reactions from the fractional factorial analysis. -

EPOXY CURING AGENTS Product Overview Guide
EPOXY CURING AGENTS Product Overview Guide NORTH AMERICA EPOXY CURING AGENTS A Robust Toolbox to Meet Diverse Formulation and End Use Requirements With a heritage built on more than 50 continuous years in the industry, Dow is the reliable leader in innovative epoxy product and technology solutions. Dow offers a robust portfolio of epoxy products, including a diverse range of curing agent chemistries and individual grades, to help meet specific formulating and application requirements. To meet increased demand for environmentally responsible formulations, our line also includes low emission and waterborne curing agents. 2 Dow leadership in epoxy technology is demonstrated by innovative technology, high quality products and attention to customer service. In addition to providing one of the broadest epoxy offerings today, our epoxy innovation pipeline – part of Dow’s $1.6 billion annual investment in research & development – ensures that the epoxy products you will require tomorrow are being created by Dow today. Beyond technology development, we are also a source of technical support to help you apply our solutions effectively so customers achieve desired results. Our comprehensive service model – which combines the Dow epoxy sales organization and our R&D and Technical Service Center in Freeport, Texas – gives you access to local know-how and support and world-class technical resources. Our commitment to the epoxy industry includes adhering to the highest standards for safety in the manufacture, storage, transport, use and disposal of our products. This guide presents a comprehensive listing of curing agents available from Dow. For assistance in choosing the right product for your formulation, or to investigate emerging innovation, please contact us. -

Dendrimer-Functionalized Hybrid Materials Based on Silica As Novel Carriers of Bioactive Acids
molecules Article Dendrimer-Functionalized Hybrid Materials Based on Silica as Novel Carriers of Bioactive Acids Mateusz Pawlaczyk * and Grzegorz Schroeder Faculty of Chemistry, Adam Mickiewicz University in Pozna´n,Uniwersytetu Pozna´nskiego 8, 61-614 Pozna´n, Poland; [email protected] * Correspondence: [email protected]; Tel.: +48-61-829-17-97 Received: 9 May 2020; Accepted: 3 June 2020; Published: 8 June 2020 Abstract: One of the major goals in the materials science is the design and development of non-toxic, versatile, and efficient drug delivery systems. The study reported in this paper concerns the syntheses of poly(amidoamine) (PAMAM) dendrimers with tris(2-aminoethyl)amine as an amine core and different terminal amines, and their attachment to silica matrix. The obtained ethylenediamine (EDA), triethylenetetramine (TETA), tris(2-aminoethyl)amine (TREN) and 4,7,10-trioxa-1,13-tridecanediamine (TRI-OXA) dendrimers were introduced to the support surface via an epoxy linker, leading to a loading 1 efficiency in the range of 0.054–0.113 mmol g− , determined using elemental and thermogravimetric analyses. The materials exhibited high adsorption capacities towards the chosen model drugs: folic, salicylic and nicotinic acid. The investigated adsorption processes were found to follow the Freundlich isotherm model, with indication of the drugs’ structure influence on the binding efficiency. Drug-loaded hybrid materials were also described for in vitro drug release in three pH-different paraphysiological media. The highest percentage release was obtained in the tests performed at pH 2.0, ranging between 35.42 and 99.83%. Satisfactory results and the versatility of PAMAM dendrimers may lead to the application of such materials not only as drug carriers dedicated to a wide range of pharmaceutics, but also as analytical tools for pre-concentration and/or the determination of biocompound contamination in samples. -

Tetraethylenepentamine Cas N°: 112-57-2
OECD SIDS TETRAETHYLENEPENTAMINE FOREWORD INTRODUCTION TETRAETHYLENEPENTAMINE CAS N°: 112-57-2 UNEP PUBLICATIONS 1 OECD SIDS TETRAETHYLENEPENTAMINE SIDS Initial Assessment Report For 13th SIAM (Bern, Switzerland November 6-9, 2001) Chemical Name: Tetraethylenepentamine CAS No.: 112-57-2 Sponsor Country: USA SIDS Contact: Oscar Hernandez United States Environmental Protection Agency (7403M) ICC Building, room 6220A 1200 Pennsylvania Avenue, NW Washington, DC 20460 History: Tetraethylenepentamine was volunteered for the U.S. HPV program and subsequently the ICCA program by the Ethyleneamines Product Stewardship Discussion Group in the U.S. Use of data from the analog triethylenetetramine is proposed to reduce testing needs. The panel/consortia (Dow, UCC and Azko-Nobel) searched company files and publicly available databases to obtain data on TEPA. The “Environmental Risk Assessment of Complexing Agents” submitted by Germany was also included in this evaluation. Testing: No testing (x) Testing ( ) Comments: Deadline for Circulation: September 14, 2001 Date of Circulation: 2 UNEP Publications OECD SIDS TETRAETHYLENEPENTAMINE SIDS INITIAL ASSESSMENT PROFILE CAS No. 112-57-2 Chemical Name 3,6,9-triazaundecamethylenediamine; tetraethylenepentamine (TEPA) Structural Formula NH2CH2CH2NHCH2CH2NHCH2CH2NHCH2CH2NH2 RECOMMENDATIONS The chemical is currently of low priority for further work. SUMMARY CONCLUSIONS OF THE SIAR Use of Analog TETA to suppleme nt TEPA data Tetraethylenepentamine (TEPA) is similar toxicologically to triethylenetetramine (TETA) based on its structure and chelation properties. Therefore, data obtained using TETA have been used to address the endpoints for reproductive and developmental toxicity. Human Health Tetraethylenepentamine (TEPA) has a low acute toxicity when administered orally to rats (LD50 = 3250 mg/kg). In an acute inhalation toxicity study with saturated vapor and whole body exposure, the LC50 was calculated to be >9.9 ppm (highest dose tested). -
![METHOD FINDER (Key on Page MF!34]](https://docslib.b-cdn.net/cover/8210/method-finder-key-on-page-mf-34-2618210.webp)
METHOD FINDER (Key on Page MF!34]
METHOD FINDER (Key on page MF!34] Meth Flow Rate Vol. (L) Extraction Analytical Chemical No. Method Name (L/min) Technique Sampler min max Acenaphthene 5506 PO LYN UC LEAR ARO MATIC 2.0 200 - 1000 aceto nitrile HPLC!FL/UV PTFE & XAD!2 HYDROCARBO NS by HPLC Acenaphthene 5515 PO LYN UC LEAR ARO MATIC 2.0 200 - 1000 varies GC!FID PTFE & XAD!2 HYDROCARBONS Acenaphthylene 5506 PO LYN UC LEAR ARO MATIC 2.0 200 - 1000 aceto nitrile HPLC!FL/UV PTFE & XAD!2 HYDROCARBO NS by HPLC Acenaphthylene 5515 PO LYN UC LEAR ARO MATIC 2.0 200 ! 1000 varies GC!FID PTFE & XAD!2 HYDROCARBONS Acetaldehyde 2018 ALIPHATIC ALDEHYDES 0.1 - 1.5 1 - 15 ACN HPLC/UV SG-DNPH Acetaldehyde 2538 ACETALDEHYDE 0.01 - 0.05 1 ! 12 tol GC!FID XAD!2/HMP Acetaldehyde 2539 ALDEHYDES, SCREENING 0.01 - 0.05 5 - 5 tol GC!FID/MS XAD-2/HMP Acetaldehyde 3507 ACETALDEHYDE 0.1 - 0.5 6 - 60 reag HPLC!UV BuB Ac etic acid 1603 AC ETIC AC ID 0.01 - 0.1 20 - 300 formic a GC!FID CCT Acetic anhydride 3506 ACETIC ANHYDRIDE 0.2 - 1.0 25 - 100 reag VIS BuB Ac eto in 2558 AC ETO IN 0.01 - 0.2 1 - 10 Acet-Meth GC-FID A-CMS Acetone 1300 KETONES I 0.01 - 0.2 0.5 - 3 CS2 GC!FID CCT Acetone 2549 VOLATILE ORGANIC CPDS 0.01 - 0.05 1 - 6 thermal TD/GC!MS TD (Screening) Acetone 3800 ORGANIC AND INORGANIC GASES 0.1 - 2.0 Instr. -

Chemicals Subject to TSCA Section 12(B) Export Notification Requirements (January 16, 2020)
Chemicals Subject to TSCA Section 12(b) Export Notification Requirements (January 16, 2020) All of the chemical substances appearing on this list are subject to the Toxic Substances Control Act (TSCA) section 12(b) export notification requirements delineated at 40 CFR part 707, subpart D. The chemicals in the following tables are listed under three (3) sections: Substances to be reported by Notification Name; Substances to be reported by Mixture and Notification Name; and Category Tables. TSCA Regulatory Actions Triggering Section 12(b) Export Notification TSCA section 12(b) requires any person who exports or intends to export a chemical substance or mixture to notify the Environmental Protection Agency (EPA) of such exportation if any of the following actions have been taken under TSCA with respect to that chemical substance or mixture: (1) data are required under section 4 or 5(b), (2) an order has been issued under section 5, (3) a rule has been proposed or promulgated under section 5 or 6, or (4) an action is pending, or relief has been granted under section 5 or 7. Other Section 12(b) Export Notification Considerations The following additional provisions are included in the Agency's regulations implementing section 12(b) of TSCA (i.e. 40 CFR part 707, subpart D): (a) No notice of export will be required for articles, except PCB articles, unless the Agency so requires in the context of individual section 5, 6, or 7 actions. (b) Any person who exports or intends to export polychlorinated biphenyls (PCBs) or PCB articles, for any purpose other than disposal, shall notify EPA of such intent or exportation under section 12(b). -
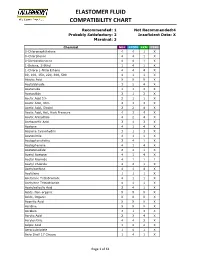
Elastomer Fluid Compatibility Chart
ELASTOMER FLUID COMPATIBILITY CHART Recommended: 1 Not Recommended:4 Probably Satisfactory: 2 Insuficient Data: X Marginal: 3 Chemical NBR EPDM FKM PTFE 0-Chloronaphthalene 4 4 1 X 0-Chlorphenol 4 4 1 X 0-Dichlorobenzene 4 4 1 X 1-Butene, 2-Ethyl 1 4 1 X 1-Chloro 1-Nitro Ethane 4 4 4 X 90, 100, 150, 220, 300, 500 4 1 1 X Abietic Acid X X X X Acetaldehyde 3 2 4 X Acetamide 1 1 3 X Acetanilide 3 1 3 X Acetic Acid 5% 2 1 1 X Acetic Acid, 30% 2 1 3 X Acetic Acid, Glacial 2 2 4 X Acetic Acid, Hot, High Pressure 4 3 4 X Acetic Anhydride 4 2 4 X Acetoacetic Acid 3 1 3 X Acetone 4 1 4 X Acetone Cyanohydrin 3 1 3 X Acetonitrile 3 1 1 X Acetophenetidine 2 4 1 X Acetophenone 4 1 4 X Acetotoluidide 2 4 1 X Acetyl Acetone 4 1 4 X Acetyl Bromide 4 1 1 1 Acetyl Chloride 4 4 1 X Acetylacetone 4 1 4 X Acetylene 1 1 1 X Acetylene Tetrabromide 4 1 1 X Acetylene Tetrachloride 4 1 1 X Acetylsalicylic Acid 2 4 1 X Acids, Non-organic X X X X Acids, Organic X X X X Aconitic Acid X X X X Acridine X X X X Acrolein 3 1 3 X Acrylic Acid 2 3 4 X Acrylonitrile 4 4 3 X Adipic Acid 1 2 2 X Aero Lubriplate 1 4 1 X Aero Shell 17 Grease 1 4 1 X Page 1 of 61 ELASTOMER FLUID COMPATIBILITY CHART Recommended: 1 Not Recommended:4 Probably Satisfactory: 2 Insuficient Data: X Marginal: 3 Chemical NBR EPDM FKM PTFE Aero Shell 1AC Grease 1 4 1 X Aero Shell 750 2 4 1 X Aero Shell 7A Grease 1 4 1 X Aerosafe 2300 4 1 4 X Aerosafe 2300W 4 1 4 X Aerozene 50 (50% Hydrazine 50% UDMH) 3 1 4 X Air, 1 1 1 X Air, 200 - 300° F 3 2 1 X Air, 300 - 400° F 4 4 1 X Air, 400 - 500° F 4 4 3 X Air, -

Study of Potential Impact of Hydraulic Fracturing on Drinking Water
EPA 601/R-12/011 | December 2012 | www.epa.gov/hfstudy Study of the Potential Impacts of Hydraulic Fracturing on Drinking Water Resources PROGRESS REPORT United States Environmental Protection Agency Office of Research and Development this page intentially left blank Study of the Potential Impacts of Hydraulic Fracturing on Drinking Water Resources PROGRESS REPORT US Environmental Protection Agency Office of Research and Development Washington, DC December 2012 EPA/601/R-12/011 Study of the Potential Impacts of Hydraulic Fracturing on Drinking Water Resources: Progress Report December 2012 Disclaimer Mention of trade names or commercial products does not constitute endorsement or recommendation for use. ii Study of the Potential Impacts of Hydraulic Fracturing on Drinking Water Resources: Progress Report December 2012 Table of Contents Executive Summary .................................................................................................................................... 1 1. Introduction ......................................................................................................................................... 5 1.1. Stakeholder Engagement ...................................................................................................... 6 2. Overview of the Research Program .................................................................................................. 8 2.1. Research Questions ........................................................................................................... -

Gad Compounds Assessed
Name CAS # ((5-Amino-2-carboxyphenyl)thio)gold sodium salt 17010-23-0 (2-Hydroxypropyl)-γ-cyclodextrin 128446-34-4 (4-hydroxy-4-isobutylpiperidin-1-yl)(1H-imidazol-1-yl)methanone (5aR, 9aR)-octahydrobenzo[e][1,3,2]dioxathiepine 3,3- (LMPC) 1-myristoyl-2-hydroxy-sn-glycero-3-phosphocholine 20559-16-4 (Trimethylsilyl)acetic acid 2345-38-2 (Z) 7-Hexadecenal 56797-40-1 (Z)-2-chloro-4-(diethylamino)but-2-en-1-yl 2-cyclohexyl-2-hydroxy-2-phenylacetate (Z)-3-chloro-4-(diethylamino)but-2-en-1-yl 2-cyclohexyl-2-hydroxy-2-phenylacetate (Z,Z,Z) 8,11,14-Eicosatrienoic acid 1783-84-2 1-(2-butoxyethoxy)-ethanol 5446-78-5 1,1,1,3,5,5,7,7,7-Nonamethyl-3-(trimethylsiloxy) tetrasiloxane 38146-99-5 1,10-decanedithiol 1191-67-9 1,1'-bicyclohexyl-4,4'-diol 20601-38-1 1,1-bis(p-ethoxyphenyl)-2,2-dimethylpropane 27955-87-9 1,1-carbonyldiimidazole (CDI) 530-62-1 1,1-cyclohexanedithiol 3855-24-1 1,2,3,4-Tetrahydronaphthalene 119-6-2 1,2,3-trichloro-2-methylpropane 1871-58-5 1,2,3-triethylbenzene 42205-08-3 1,2,4-benzenetriol 533-73-3 1,2,4-triethylbenzene 877-44-1 1,2-cyclohexanediamine 694-83-7 1,2-cyclohexanedicarboxylic acid 1687-30-5 1,2-cyclohexanediol 931-17-9 1,2-dichlorobenzene 95-50-1 1,2-Ethanediamine 107-15-3 1,2-ethanedithiol 540-63-6 1,2-propanediol, monobutyl ether 29387-86-8 1,3,5-Tributyl-2,4,6-trioxo-hexahydro-striazine 846-74-2 1,3,5-triethylbenzene 102-25-0 1,3,5-Trioxane 110-88-3 1,3:2,4-Bis(3,4-dimethylbenzylidene)sorbitol 135861-56-2 1,3-Bis(diallylamino)-2-propanol 4831-46-3 1,3-butanediol 107-88-0 1,3-cycloheptadiene 4054-38-0 1,3-cyclohexanedicarboxylic -

Triethylenetetramine
Triethylenetetramine sc-251328 Material Safety Data Sheet Hazard Alert Code Key: EXTREME HIGH MODERATE LOW Section 1 - CHEMICAL PRODUCT AND COMPANY IDENTIFICATION PRODUCT NAME Triethylenetetramine STATEMENT OF HAZARDOUS NATURE CONSIDERED A HAZARDOUS SUBSTANCE ACCORDING TO OSHA 29 CFR 1910.1200. NFPA FLAMMABILITY1 HEALTH3 HAZARD INSTABILITY0 SUPPLIER Santa Cruz Biotechnology, Inc. 2145 Delaware Avenue Santa Cruz, California 95060 800.457.3801 or 831.457.3800 EMERGENCY ChemWatch Within the US & Canada: 877-715-9305 Outside the US & Canada: +800 2436 2255 (1-800-CHEMCALL) or call +613 9573 3112 SYNONYMS C6-H18-N4, (H2NCH2CH2NHCH2)2, H2N(C2-H2-NH)3H, NH2(CH2)2NH(CH2)2NH(CH2)2NH2, "N, N' -bis(2-aminoethyl)-1, 2-diaminoethane", "1, 8-diamino-3, 6-diazooctane", "N, N' -bis(2-aminoethyl) ethylenediamine", "3, 6-diazaoctane-1, 8-diamine", tetralin, "1, 2-ethanediamine, N, N' -bis(2-aminoethyl)-", "ethylenediamine, N, N' -bis(2-aminoethyl)-", "1, 4, 7, 10-tetraazadecane", "Araldite Hardener HY 951", TETA, "DEH 24", "Araldite HY 951", TEZCA, "GE Material D5F15B1", "Hysol H2-3404", "Ludowici Wear Resist Trowelable (Part B)", Trien, Trientine Section 2 - HAZARDS IDENTIFICATION CHEMWATCH HAZARD RATINGS Min Max Flammability: 1 Toxicity: 2 Body Contact: 3 Min/Nil=0 Low=1 Reactivity: 1 Moderate=2 High=3 Chronic: 3 Extreme=4 1 of 9 CANADIAN WHMIS SYMBOLS EMERGENCY OVERVIEW RISK Harmful in contact with skin. Causes burns. Risk of serious damage to eyes. May cause SENSITISATION by skin contact. Harmful to aquatic organisms, may cause long-term adverse effects in the aquatic environment. POTENTIAL HEALTH EFFECTS ACUTE HEALTH EFFECTS SWALLOWED ! The material can produce chemical burns within the oral cavity and gastrointestinal tract following ingestion.