On the Construction of Binary Mixture P-X and T-X Diagrams from Isochoric
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Phase Transitions in Multicomponent Systems
Physics 127b: Statistical Mechanics Phase Transitions in Multicomponent Systems The Gibbs Phase Rule Consider a system with n components (different types of molecules) with r phases in equilibrium. The state of each phase is defined by P,T and then (n − 1) concentration variables in each phase. The phase equilibrium at given P,T is defined by the equality of n chemical potentials between the r phases. Thus there are n(r − 1) constraints on (n − 1)r + 2 variables. This gives the Gibbs phase rule for the number of degrees of freedom f f = 2 + n − r A Simple Model of a Binary Mixture Consider a condensed phase (liquid or solid). As an estimate of the coordination number (number of nearest neighbors) think of a cubic arrangement in d dimensions giving a coordination number 2d. Suppose there are a total of N molecules, with fraction xB of type B and xA = 1 − xB of type A. In the mixture we assume a completely random arrangement of A and B. We just consider “bond” contributions to the internal energy U, given by εAA for A − A nearest neighbors, εBB for B − B nearest neighbors, and εAB for A − B nearest neighbors. We neglect other contributions to the internal energy (or suppose them unchanged between phases, etc.). Simple counting gives the internal energy of the mixture 2 2 U = Nd(xAεAA + 2xAxBεAB + xBεBB) = Nd{εAA(1 − xB) + εBBxB + [εAB − (εAA + εBB)/2]2xB(1 − xB)} The first two terms in the second expression are just the internal energy of the unmixed A and B, and so the second term, depending on εmix = εAB − (εAA + εBB)/2 can be though of as the energy of mixing. -

Grade 6 Science Mechanical Mixtures Suspensions
Grade 6 Science Week of November 16 – November 20 Heterogeneous Mixtures Mechanical Mixtures Mechanical mixtures have two or more particle types that are not mixed evenly and can be seen as different kinds of matter in the mixture. Obvious examples of mechanical mixtures are chocolate chip cookies, granola and pepperoni pizza. Less obvious examples might be beach sand (various minerals, shells, bacteria, plankton, seaweed and much more) or concrete (sand gravel, cement, water). Mechanical mixtures are all around you all the time. Can you identify any more right now? Suspensions Suspensions are mixtures that have solid or liquid particles scattered around in a liquid or gas. Common examples of suspensions are raw milk, salad dressing, fresh squeezed orange juice and muddy water. If left undisturbed the solids or liquids that are in the suspension may settle out and form layers. You may have seen this layering in salad dressing that you need to shake up before using them. After a rain fall the more dense particles in a mud puddle may settle to the bottom. Milk that is fresh from the cow will naturally separate with the cream rising to the top. Homogenization breaks up the fat molecules of the cream into particles small enough to stay suspended and this stable mixture is now a colloid. We will look at colloids next. Solution, Suspension, and Colloid: https://youtu.be/XEAiLm2zuvc Colloids Colloids: https://youtu.be/MPortFIqgbo Colloids are two phase mixtures. Having two phases means colloids have particles of a solid, liquid or gas dispersed in a continuous phase of another solid, liquid, or gas. -

Introduction to Phase Diagrams*
ASM Handbook, Volume 3, Alloy Phase Diagrams Copyright # 2016 ASM InternationalW H. Okamoto, M.E. Schlesinger and E.M. Mueller, editors All rights reserved asminternational.org Introduction to Phase Diagrams* IN MATERIALS SCIENCE, a phase is a a system with varying composition of two com- Nevertheless, phase diagrams are instrumental physically homogeneous state of matter with a ponents. While other extensive and intensive in predicting phase transformations and their given chemical composition and arrangement properties influence the phase structure, materi- resulting microstructures. True equilibrium is, of atoms. The simplest examples are the three als scientists typically hold these properties con- of course, rarely attained by metals and alloys states of matter (solid, liquid, or gas) of a pure stant for practical ease of use and interpretation. in the course of ordinary manufacture and appli- element. The solid, liquid, and gas states of a Phase diagrams are usually constructed with a cation. Rates of heating and cooling are usually pure element obviously have the same chemical constant pressure of one atmosphere. too fast, times of heat treatment too short, and composition, but each phase is obviously distinct Phase diagrams are useful graphical representa- phase changes too sluggish for the ultimate equi- physically due to differences in the bonding and tions that show the phases in equilibrium present librium state to be reached. However, any change arrangement of atoms. in the system at various specified compositions, that does occur must constitute an adjustment Some pure elements (such as iron and tita- temperatures, and pressures. It should be recog- toward equilibrium. Hence, the direction of nium) are also allotropic, which means that the nized that phase diagrams represent equilibrium change can be ascertained from the phase dia- crystal structure of the solid phase changes with conditions for an alloy, which means that very gram, and a wealth of experience is available to temperature and pressure. -

Gp-Cpc-01 Units – Composition – Basic Ideas
GP-CPC-01 UNITS – BASIC IDEAS – COMPOSITION 11-06-2020 Prof.G.Prabhakar Chem Engg, SVU GP-CPC-01 UNITS – CONVERSION (1) ➢ A two term system is followed. A base unit is chosen and the number of base units that represent the quantity is added ahead of the base unit. Number Base unit Eg : 2 kg, 4 meters , 60 seconds ➢ Manipulations Possible : • If the nature & base unit are the same, direct addition / subtraction is permitted 2 m + 4 m = 6m ; 5 kg – 2.5 kg = 2.5 kg • If the nature is the same but the base unit is different , say, 1 m + 10 c m both m and the cm are length units but do not represent identical quantity, Equivalence considered 2 options are available. 1 m is equivalent to 100 cm So, 100 cm + 10 cm = 110 cm 0.01 m is equivalent to 1 cm 1 m + 10 (0.01) m = 1. 1 m • If the nature of the quantity is different, addition / subtraction is NOT possible. Factors used to check equivalence are known as Conversion Factors. GP-CPC-01 UNITS – CONVERSION (2) • For multiplication / division, there are no such restrictions. They give rise to a set called derived units Even if there is divergence in the nature, multiplication / division can be carried out. Eg : Velocity ( length divided by time ) Mass flow rate (Mass divided by time) Mass Flux ( Mass divided by area (Length 2) – time). Force (Mass * Acceleration = Mass * Length / time 2) In derived units, each unit is to be individually converted to suit the requirement Density = 500 kg / m3 . -

Partition Coefficients in Mixed Surfactant Systems
Partition coefficients in mixed surfactant systems Application of multicomponent surfactant solutions in separation processes Vom Promotionsausschuss der Technischen Universität Hamburg-Harburg zur Erlangung des akademischen Grades Doktor-Ingenieur genehmigte Dissertation von Tanja Mehling aus Lohr am Main 2013 Gutachter 1. Gutachterin: Prof. Dr.-Ing. Irina Smirnova 2. Gutachterin: Prof. Dr. Gabriele Sadowski Prüfungsausschussvorsitzender Prof. Dr. Raimund Horn Tag der mündlichen Prüfung 20. Dezember 2013 ISBN 978-3-86247-433-2 URN urn:nbn:de:gbv:830-tubdok-12592 Danksagung Diese Arbeit entstand im Rahmen meiner Tätigkeit als wissenschaftliche Mitarbeiterin am Institut für Thermische Verfahrenstechnik an der TU Hamburg-Harburg. Diese Zeit wird mir immer in guter Erinnerung bleiben. Deshalb möchte ich ganz besonders Frau Professor Dr. Irina Smirnova für die unermüdliche Unterstützung danken. Vielen Dank für das entgegengebrachte Vertrauen, die stets offene Tür, die gute Atmosphäre und die angenehme Zusammenarbeit in Erlangen und in Hamburg. Frau Professor Dr. Gabriele Sadowski danke ich für das Interesse an der Arbeit und die Begutachtung der Dissertation, Herrn Professor Horn für die freundliche Übernahme des Prüfungsvorsitzes. Weiterhin geht mein Dank an das Nestlé Research Center, Lausanne, im Besonderen an Herrn Dr. Ulrich Bobe für die ausgezeichnete Zusammenarbeit und der Bereitstellung von LPC. Den Studenten, die im Rahmen ihrer Abschlussarbeit einen wertvollen Beitrag zu dieser Arbeit geleistet haben, möchte ich herzlichst danken. Für den außergewöhnlichen Einsatz und die angenehme Zusammenarbeit bedanke ich mich besonders bei Linda Kloß, Annette Zewuhn, Dierk Claus, Pierre Bräuer, Heike Mushardt, Zaineb Doggaz und Vanya Omaynikova. Für die freundliche Arbeitsatmosphäre, erfrischenden Kaffeepausen und hilfreichen Gespräche am Institut danke ich meinen Kollegen Carlos, Carsten, Christian, Mohammad, Krishan, Pavel, Raman, René und Sucre. -

Ways to Physically Separate a Mixture There Are 2 Types of Mixtures
Mixtures 12 Mixtures Homogenous Compare Heterogeneous compounds and Suspension Mixtures. Colloid Solution Differentiate Solute/Solvent between solutions, suspensions, and colloids Comparing Mixtures and Compounds Mixtures Compounds Made of 2 or more substances Made of 2 or more substances physically combined chemically combined Substances keep their own properties Substances lose their own properties Can be separated by physical means Can be separated only by chemical means Have no definite chemical composition Have a definite chemical composition What method is used to separate mixtures Ways to physically separate a Mixture based on boiling • Distillation – uses boiling point point? • Magnet – uses magnetism What method is used • Centrifuge – uses density to separate mixtures based on density? • Filtering – separates large particles from smaller ones What method is used There are 2 types of mixtures: to separate mixtures (1) Heterogeneous Mixtures: the parts mixed together can still be based on particle size? distinguished from one another...NOT uniform in composition Give examples of a heterogeneous Examples: chicken soup, fruit salad, dirt, sand in water mixture Mixtures 12 (2) Homogenous Mixtures: the parts mixed together cannot be distinguished from one another...completely uniform in composition. Give examples of a homogenous Examples: Air, Kool-aid, Brass, salt water, milk mixture Differentiate Types of Homogenous mixtures between a homogenous 1. Suspensions mixture and a i.e. chocolate milk, muddy water, Italian dressing heterogeneous mixture. They are cloudy (usually a liquid mixed with small solid particles) Identify an example of a suspension. Needs to be shaken or stirred to keep the solids from Will the solid settling particles settle in a suspension? The solids can be filtered out 2. -

Solving Mixture and Solution Verbal Problems
SOLVING SOLUTION AND MIXTURE VERBAL PROBLEMS This type of problem involves mixing two different solutions of a certain ingredient to get a desired concentration of the ingredient. Before we can solve problems that involve concentrations, we must review certain concepts about percents. If you need to do this, go to the brush-up materials for solving percent problems on the Dolciani website. 1. Solution Problems Basic Equation: amount of solution concentration of substance = amount of substance Example: 40 ounces (amount of solution) of a 25% solution of acid (concentration) contains 25(40) = 10 ounces of acid Usual equation to solve for the variable: Amount of substance in solution 1 + Amount of substance in solution 2 = Amount of substance in solution 2. Mixture Problems Basic Equation: unit price # units = cost (or value) Example: 5 pounds of apples (# units) that sell for $1.20 per pound (unit price) costs 5(1.20) = $6 Using equation to solve for the variable: Cost of ingredient 1 + Cost of ingredient 2 = Cost of mixture Now let’s look at an actual mixture problem. It is easiest when solving a mixture problem to make a table to get the information organized. No matter the story line of the problem, the table can be used and labelled as necessary. Let’s look at a few examples. Example 1: Fatima’s chemistry lab stocks an 8% acid solution and a 20% acid solution. How many ounces of each must she combine to produce 60 ounces of a mixture that is 10% acid? Solution: We want to find how much of each solution must be in the mixture. -

Mixtures Are a Combination 3.1 of Two Or More Substances MIXTURES
Mixtures are a combination 3.1 of two or more substances MIXTURES A solution is a solute dissolved 3.2 in a solvent Mixtures can be separated according 3.3 to their physical properties 3.4 Mixtures can be separated according 3 to their size and mass What if? The different boiling points of liquids Case mix 3.5 can be used to separate mixtures What you need: a variety of different pencil cases (size, shape, colour) What to do: 1 Place all the pencil cases in Solubility can be used to one pile. 3.6 separate mixtures 2 List your pencil case’s properties that will allow it to DRAFT be identified easily (e.g. colour, shape, size and weight). 3 Give the list to another student. Can they identify your case by Waste water is a mixture that using the list? 3.7 can be separated What if? » What if you were blindfolded? Could you still find your pencil case? » What if the pencil cases were too small to feel? How could Materials recovery facilities you identify yours? 3.8 separate mixtures » What if all the pencil cases were exactly the same? Would it still be a mixture? Mixtures are a 3.1 combination of two or more substances Consider the things around you. Perhaps they are made of wood, glass or plastic. Wood, glass and plastic are all mixtures – each of these materials is made up of two or more substances. Some materials are pure substances. A pure substance is one where all the particles are identical. -

Classification of Matter Section ●1 Composition of Matter
266_277_Ch15_RE_896315.qxd 3/23/10 3:04 AM Page 266 S-034 113:GO00492:GPS_Reading_Essentials_SE%0:XXXXXXXXXXXXX_SE:Application_Files_ chapter 15 Classification of Matter section ●1 Composition of Matter What You’ll Learn Before You Read ■ what substances and mixtures are Matter is all around you. You breathe matter, sit on it, and ■ how to identify drink it every day. What words would you use to describe elements and different kinds of matter? compounds ■ the difference between solutions, colloids, and suspensions Read to Learn Underline Look for different descriptions of matter Pure Substances as you read each paragraph. Underline these descriptions. Have you ever seen a print that looked like a real painting? Read the underlined descriptions Did you have to touch it to find out? The smooth or rough again after you’ve finished surface told you whether it was a painting or a print. Each reading the section. material has its own properties. The properties of materials can be used to classify them into categories. Each material is made of a pure substance or of a mix of substances. A substance is a type of matter that is always made of the same material or materials. A substance can be either an element or a compound. Some substances you might recognize are helium, aluminum, water, and salt. What are elements? All substances are made of atoms. A substance is an element if all the atoms in the substance are the same. The graphite in your pencil is an element. The copper coating on most pennies is an element, too. -

Solving a Solution Mixture Problem
Only to be used for arranged hours, will count as two activities Math 31 Activity # 13 “Solving Mixture Problems” Your Name: ___________________ Solving Word Problems: The Cohort Strategy! Step 1) Read the problem at least once carefully. Look for key words and phrases. Determine the known and unknown quantities. Let x or another variable to represent one of the unknown quantities in the problem. Step 2) If necessary, using the same variable from Step 1, write an expression using the to represent any other unknown quantities. Step 3) Write a summary of the problem as an English statement. Then write an equation based on your summary. Step 4) Solve the equation. Step 5) Check the solution. Ask yourself "Is my answer reasonable?" Step 6) Write a sentence to state what was asked for in the problem, and be sure to include units as part of the solution. (inches, square feet, gallons, ounces, etc., for example). Only to be used for arranged hours, will count as two activities Task 1) Percent Mixture Problems Example 1: How many gallons of a 15% sugar solution must be mixed with 5 gallons of a 40% sugar solution to make a 30% sugar solution? Begin by creating a drawing of the situation and filling in the known information, as shown below. Amount of Solution + ___ gallons = + = Concentration ___ ____ ____ (Percents) Amount of Ingredient in Mixture + = Now we must decide how to represent the unknown quantities. Since the question is asking us to find the number of gallons of 15% solution we will use to obtain the required result, it will be useful to have our variable represent the amount of 15% sugar solution we are using. -
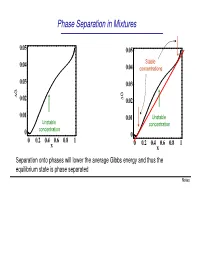
Phase Separation in Mixtures
Phase Separation in Mixtures 0.05 0.05 0.04 Stable 0.04 concentrations 0.03 0.03 G G ∆ 0.02 ∆ 0.02 0.01 0.01 Unstable Unstable concentration concentration 0 0 0 0.2 0.4 0.6 0.8 1 0 0.2 0.4 0.6 0.8 1 x x Separation onto phases will lower the average Gibbs energy and thus the equilibrium state is phase separated Notes Phase Separation versus Temperature Note that at higher temperatures the region of concentrations where phase separation takes place shrinks and G eventually disappears T increases ∆G = ∆U −T∆S This is because the term -T∆S becomes large at high temperatures. You can say that entropy “wins” over the potential energy cost at high temperatures Microscopically, the kinetic energy becomes x much larger than potential at high T, and the 1 x2 molecules randomly “run around” without noticing potential energy and thus intermix Oil and water mix at high temperature Notes Liquid-Gas Phase Separation in a Mixture A binary mixture can exist in liquid or gas phases. The liquid and gas phases have different Gibbs potentials as a function of mole fraction of one of the components of the mixture At high temperatures the Ggas <Gliquid because the entropy of gas is greater than that of liquid Tb2 T At lower temperatures, the two Gibbs b1 potentials intersect and separation onto gas and liquid takes place. Notes Boiling of a binary mixture Bubble point curve Dew point If we have a liquid at pint A that contains F A gas curve a molar fraction x1 of component 1 and T start heating it: E D - First the liquid heats to a temperature T’ C at constant x A and arrives to point B. -

Title: What Is a Mixture? Objective: to Help Students Understand That
Title: What is a Mixture? Objective: To help students understand that mixtures are two or more pure substances that are not combined chemically. This means that they can be separated back into their respective pure substances. Standards: EALR 4 Physical Science: Big Idea: Matter: Properties and Change. Materials: Box of Fruit Loops, Box of Corn Flakes, Box of Raisins, quart sized wide mouth jar or gallon sized clear storage bag. Procedure: Show students 2 cups of Fruit Loops in a quart jar or plastic bag. Hold up the box of corn Flakes and the box of raisins. Discuss question #1. Point out that a mixture is a combination of substances. Pour 1 cup of corn flakes into the fruit loops. Gently mix the two substances. Hold the jar or bag up so students may observe. Discuss question #2. Pour ½ cup of raisins into the mixture of fruit loops and corn flakes. Gently mix the raisins into the mixture. Hold the jar or bag up so students may observe. Explain that a mixture is a combination of two or more substances not chemically combined. Pour the fruit loop, corn flake, and raisin mixture onto a clean sheet of paper. Discuss question #3. Student Questions for Inquiry: 1. What kind of substances are fruit loops, corn flakes, and raisins? (They are all solids.) 2. What kind of matter do we have now? (The students should respond by saying that it is a mixture.) 3. How could we separate the fruit loops from the corn flakes and raisins? The raisins from the corn flakes? (The respective ingredients can be easily picked out of the mixture.) Science Behind It: A mixture is a substance made by combining two or more different materials in such a way that no chemical reaction occurs.