AIS Laboratory Standard Operating Procedures
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-
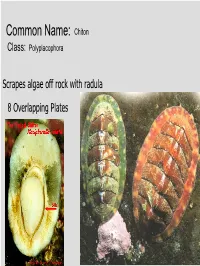
Common Name: Chiton Class: Polyplacophora
Common Name: Chiton Class: Polyplacophora Scrapes algae off rock with radula 8 Overlapping Plates Phylum? Mollusca Class? Gastropoda Common name? Brown sea hare Class? Scaphopoda Common name? Tooth shell or tusk shell Mud Tentacle Foot Class? Gastropoda Common name? Limpet Phylum? Mollusca Class? Bivalvia Class? Gastropoda Common name? Brown sea hare Phylum? Mollusca Class? Gastropoda Common name? Nudibranch Class? Cephalopoda Cuttlefish Octopus Squid Nautilus Phylum? Mollusca Class? Gastropoda Most Bivalves are Filter Feeders A B E D C • A: Mantle • B: Gill • C: Mantle • D: Foot • E: Posterior adductor muscle I.D. Green: Foot I.D. Red Gills Three Body Regions 1. Head – Foot 2. Visceral Mass 3. Mantle A B C D • A: Radula • B: Mantle • C: Mouth • D: Foot What are these? Snail Radulas Dorsal HingeA Growth line UmboB (Anterior) Ventral ByssalC threads Mussel – View of Outer Shell • A: Hinge • B: Umbo • C: Byssal threads Internal Anatomy of the Bay Mussel A B C D • A: Labial palps • B: Mantle • C: Foot • D: Byssal threads NacreousB layer Posterior adductorC PeriostracumA muscle SiphonD Mantle Byssal threads E Internal Anatomy of the Bay Mussel • A: Periostracum • B: Nacreous layer • C: Posterior adductor muscle • D: Siphon • E: Mantle Byssal gland Mantle Gill Foot Labial palp Mantle Byssal threads Gill Byssal gland Mantle Foot Incurrent siphon Byssal Labial palp threads C D B A E • A: Foot • B: Gills • C: Posterior adductor muscle • D: Excurrent siphon • E: Incurrent siphon Heart G F H E D A B C • A: Foot • B: Gills • C: Mantle • D: Excurrent siphon • E: Incurrent siphon • F: Posterior adductor muscle • G: Labial palps • H: Anterior adductor muscle Siphon or 1. -

Greening Phenomenon in Bivalve by Marennine Produced from Haslea Ostrearia and Its Consequences on Bivalve's Integrated Resp
Greening phenomenon in bivalve by marennine produced from Haslea ostrearia and its consequences on bivalve’s integrated response Fiddy Semba Prasetiya To cite this version: Fiddy Semba Prasetiya. Greening phenomenon in bivalve by marennine produced from Haslea os- trearia and its consequences on bivalve’s integrated response. Invertebrate Zoology. Université du Maine, 2015. English. NNT : 2015LEMA1017. tel-01279527 HAL Id: tel-01279527 https://tel.archives-ouvertes.fr/tel-01279527 Submitted on 26 Feb 2016 HAL is a multi-disciplinary open access L’archive ouverte pluridisciplinaire HAL, est archive for the deposit and dissemination of sci- destinée au dépôt et à la diffusion de documents entific research documents, whether they are pub- scientifiques de niveau recherche, publiés ou non, lished or not. The documents may come from émanant des établissements d’enseignement et de teaching and research institutions in France or recherche français ou étrangers, des laboratoires abroad, or from public or private research centers. publics ou privés. Fiddy SEMBA PRASETIYA Mémoire présenté en vue de l’obtention du grade de Docteur de l’Université du Maine sous le label de L’Université Nantes Angers Le Mans École doctorale : Végétale Environnement Nutrition Agro-alimentaire Mer (VENAM) Discipline : BIOLOGIÉ DES ORGANISMES Unité de recherche : MER MOLÉCULE ET SANTÉ (MMS) – EA n°2160, Université du Maine, UFR Sciences et Techniques, Avenue Olivier Messiaen 72085 Le Mans Cedex 9 Soutenue le 27 Novembre 2015 Greening phenomenon in bivalve by marennine -

Silicified Eocene Molluscs from the Lower Murchison District, Southern Carnarvon Basin, Western Australia
[<ecords o{ the Western A uslralian Museum 24: 217--246 (2008). Silicified Eocene molluscs from the Lower Murchison district, Southern Carnarvon Basin, Western Australia Thomas A. Darragh1 and George W. Kendrick2.3 I Department of Invertebrate Palaeontology, Museum Victoria, 1'.0. Box 666, Melbourne, Victoria 3001, Australia. Email: tdarragh(il.Illuseum.vic.gov.au :' Department of Earth and Planetary Sciences, Western Australian Museum, Locked Bag 49, Welshpool D.C., Western Australia 6986, Australia. 1 School of Earth and Ceographical Sciences, The University of Western Australia, 35 Stirling Highway, Crawlev, Western Australia 6009, Australia. Abstract - Silicified Middle to Late Eocene shallow water sandstones outcropping in the Lower Murchison District near Kalbarri township contain a silicified fossil fauna including foraminifera, sponges, bryozoans, solitary corals, brachiopods, echinoids and molluscs. The known molluscan fauna consists of 51 species, comprising 2 cephalopods, 14 bivalves, 1 scaphopod and 34 gastropods. Of these taxa three are newly described, Cerithium lvilya, Zeacolpus bartol1i, and Lyria lamellatoplicata. 25 of these molluscs are identical to or closely comparable with taxa from the southern Australian Eocene. The occurrence of this fauna extends the Southeast Australian Province during the Eocene from southwest Western Australia along the west coast north to at least 27° present day south latitude; consequently the province is here renamed the Southern Australian Province. Keywords: siliceous fossils, Eocene, Kalbarri, molluscs, new taxa, Carnarvon Basin, biogeography, Southern Australian Province. INTRODUCTION The source deposit, a pallid to ferruginous silicified Eocene marine molluscan assemblages from sandstone, forms a weakly defined, low breakaway coastal sedimentary basins in southern Australia trending N-S and sloping gently westward. -

Larvae of Bivalve Mollusks of the Sevastopol Region of the Black Sea
W&M ScholarWorks Reports 1966 Larvae of bivalve mollusks of the Sevastopol region of the Black Sea K. A. Zakhvatkina Follow this and additional works at: https://scholarworks.wm.edu/reports Part of the Aquaculture and Fisheries Commons, Marine Biology Commons, and the Zoology Commons Recommended Citation Zakhvatkina, K. A. (1966) Larvae of bivalve mollusks of the Sevastopol region of the Black Sea. Translation series (Virginia Institute of Marine Science) ; no. 15. Virginia Institute of Marine Science, William & Mary. https://scholarworks.wm.edu/reports/39 This Report is brought to you for free and open access by W&M ScholarWorks. It has been accepted for inclusion in Reports by an authorized administrator of W&M ScholarWorks. For more information, please contact [email protected]. VIRGINIA INSTITUTE OF MARINE SCIENCE GLOUCESTER POINT, VIRGDHA .. LARVAE OF BIVALVE MOLLUSKS OF THE SEVASTOPOL REGION OF THE BLACK SEA TRANSlATION SLRIES NO· 15 1966 Virginia Institute of Marine Science Gloucester Point, Virginia URVhE OF BIVALVE hOLLUSKS OF THE SEVii.STOPOL REGION OF THE BLACK SEA By K. A· Zakhvatkina Original title: Lichinki dvustvorchatykh molliuskov sevastopol'skogo raiona Chernogo Moria From: Akademiia Nauk SSSR, Trudy Sevastopol1 skoi Biologicheskoi Stantsii, Tom XI, p• 108-152, 1959 Translated by Evelyn c. wells Edited by Paul Chanley TRANSLATION SERIES NO· 15 w. J. Hargis Director April 1966 Akad • Nauk SSSR, Trudy Sevastopol' skoi Biologicheskoi St.antsii Tom XI, P• 108-152, 1959 LJ.dtvii.E OF BIV;.,LVE HOLLUSKS. OF TH1 SLVJL.TOf'OL REGION OF THE BLACK SEA By K· A· Zakhvatkina Heretofore, the systematic relationships as well as the biology p•l08 and ecology of bivalve larvce have been poorly known• The work of A· Borisiak (1905), on the larv~e of bivalve mollusks, is of only historical interest since only four of the 20 forfus described were deter~mined to genus• Data on the reproduction of several species of bivalve mollusks, especially on spawning seasons, are given in the work of z. -
Field Identification Guide to the Living Marine Resources In
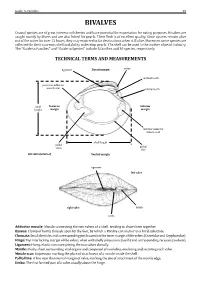
Guide to Families 29 BIVALVES Coastal species are of great interest to fisheries and have potential for exportation for eating purposes. Bivalves are caught mainly by divers and are also fished for pearls. Their flesh is of excellent quality. Since oysters remain alive out of the water for over 12 hours, they may exported to far destinations when still alive. Moreover, some species are collected for their nacreous shell and ability to develop pearls. The shell can be used in the mother of pearl industry. The “Guide to Families’’ andTECHNICAL ‘‘Guide to Species’’ TERMS include 5AND families MEASUREMENTS and 10 species, respectively. ligament Dorsal margin umbo posterior adductor cardinal tooth muscle scar lateral tooth Posterior Anterior margin margin shell height anterior adductor muscle scar pallial sinus pallial shell length line left valve (interior) Ventral margin ligament left valve right valve lunule umbo Adductor muscle: Byssus: Chomata: Muscle connecting the two valves of a shell, tending to draw them together. Hinge: Clump of horny threads spun by the foot, by which a Bivalve can anchor to a hard substrate. Ligament: Small denticles and corresponding pits located on the inner margin of the valves (Ostreidae and Gryphaeidae). Mantle: Top interlocking margin of the valves, often with shelly projections (teeth) and corresponding recesses (sockets). Muscle scar: Horny, elastic structure joining the two valves dorsally. Pallial line: Fleshy sheet surrounding vital organs and composed of two lobes, one lining and secreting each valve. Umbo: Impression marking the place of attachment of a muscle inside the shell. A line near the internal margin of valve, marking the site of attachment of the mantle edge. -

Clam Dissection Guideline
Clam Dissection Guideline BACKGROUND: Clams are bivalves, meaning that they have shells consisting of two halves, or valves. The valves are joined at the top, and the adductor muscles on each side hold the shell closed. If the adductor muscles are relaxed, the shell is pulled open by ligaments located on each side of the umbo. The clam's foot is used to dig down into the sand, and a pair of long incurrent and excurrent siphons that extrude from the clam's mantle out the side of the shell reach up to the water above (only the exit points for the siphons are shown). Clams are filter feeders. Water and food particles are drawn in through one siphon to the gills where tiny, hair-like cilia move the water, and the food is caught in mucus on the gills. From there, the food-mucus mixture is transported along a groove to the palps (mouth flaps) which push it into the clam's mouth. The second siphon carries away the water. The gills also draw oxygen from the water flow. The mantle, a thin membrane surrounding the body of the clam, secretes the shell. The oldest part of the clam shell is the umbo, and it is from the hinge area that the clam extends as it grows. I. Purpose: The purpose of this lab is to identify the internal and external structures of a mollusk by dissecting a clam. II. Materials: 2 pairs of safety goggles 1 paper towel 2 pairs of gloves 1 pair of scissors 1 preserved clam 2 pairs of forceps 1 dissecting tray 2 probes III. -

What Are Molluscs?
The shells of molluscs from all over the world – on land, in lakes, and in the ocean – contain very detailed imprints of past climate change. Using isotope analysis, we can extract these signals and start to piece together long-term climate variations. You will never look at a garden snail in the same way again! What are molluscs? Molluscs are soft-bodied (invertebrate) organisms that are widespread in terrestrial, freshwater, and marine habitats. We can split them into two basic groups: Gastropods: Molluscs with up to one shell or ‘valve’ (such as snails or slugs) Bivalves: Molluscs with two-sided shells or ‘valves’ (such as clams, oysters, and mussels) Many molluscs (both gastropods and bivalves) build hard shells that are rich in calcium carbonate. We call these ‘calcareous shells’. Such shells are formed in distinct bands (like tree rings) growing outwards along the direction of growth – the oldest shell is at the edge. Over time, more bands are added to the shell as it grows and the organism is enlarged (Figure 1a-c). The environmental conditions in which the organism live are reflected in the types of isotopes that are present in its shell. This means that the shell contains a ‘geochemical’ signature of the mollusc’s habitat and lifestyle. When the organism dies, its shell can become buried and preserved in sediments at the bottom of the lake or ocean (Figure 1d-f). Once preserved, these shells provide us with a valuable record of environmental conditions at the time that the organism lived. We can analyse the isotopes within the shells to build up a picture of environmental conditions in the past. -

Freshwater Mussels of the Pacific Northwest
Freshwater Mussels of the Pacifi c Northwest Ethan Nedeau, Allan K. Smith, and Jen Stone Freshwater Mussels of the Pacifi c Northwest CONTENTS Part One: Introduction to Mussels..................1 What Are Freshwater Mussels?...................2 Life History..............................................3 Habitat..................................................5 Role in Ecosystems....................................6 Diversity and Distribution............................9 Conservation and Management................11 Searching for Mussels.............................13 Part Two: Field Guide................................15 Key Terms.............................................16 Identifi cation Key....................................17 Floaters: Genus Anodonta.......................19 California Floater...................................24 Winged Floater.....................................26 Oregon Floater......................................28 Western Floater.....................................30 Yukon Floater........................................32 Western Pearlshell.................................34 Western Ridged Mussel..........................38 Introduced Bivalves................................41 Selected Readings.................................43 www.watertenders.org AUTHORS Ethan Nedeau, biodrawversity, www.biodrawversity.com Allan K. Smith, Pacifi c Northwest Native Freshwater Mussel Workgroup Jen Stone, U.S. Fish and Wildlife Service, Columbia River Fisheries Program Offi ce, Vancouver, WA ACKNOWLEDGEMENTS Illustrations, -

Key to the Freshwater Bivalves of New Jersey
Key to the Freshwater Bivalves of New Jersey 1. a. shell with a very sharp posterior ridge, shaped like the marine mussel, Mytilus, generally less than 30 mm, and attached to a hard substrate with byssal threads.........................……………........................Zebra mussel b. animal without byssal threads attaching adult to substrate, with or without teeth but not with the above shape................................….............................2 2. a. valves with cardinal teeth and two sets of lateral teeth.......................…...............................3 b. valves with one set of lateral teeth and pseudocardinal teeth or without teeth.............................................................................................................5 3. a. shell thick and sturdy, beak bulbous and curving anteriorly………………….Atlantic rangia b. shell moderately thick, beak not bulbous nor curving…………………………………………...4 4. a. valves with serrated lateral teeth......................................……….........................Asian clam b. valves with smooth lateral teeth....................................................................Fingernail clam 5. a. hinge teeth absent.................................................................................................................6 b. hinge teeth present..............................................................................................................10 6. a. beaks not projecting above the hinge line................…………………........ Paper pondshell b. beaks projecting above -

Mechanics Unlocks the Morphogenetic Puzzle of Interlocking Bivalved Shells
Mechanics unlocks the morphogenetic puzzle of interlocking bivalved shells Derek E. Moultona,1 , Alain Gorielya , and Regis´ Chiratb aMathematical Institute, University of Oxford, Oxford, OX2 6GG, United Kingdom; and bCNRS 5276, LGL-TPE (Le Laboratoire de Geologie´ de Lyon: Terre, Planetes,` Environnement), Universite´ Lyon 1, 69622 Villeurbanne Cedex, France Edited by Sean H. Rice, Texas Tech University, Lubbock, TX, and accepted by Editorial Board Member David Jablonski November 11, 2019 (received for review September 24, 2019) Brachiopods and mollusks are 2 shell-bearing phyla that diverged tal events causing shell injuries. Yet, in all cases the interlocking from a common shell-less ancestor more than 540 million years ago. of the 2 shell edges is tightly maintained. These observations Brachiopods and bivalve mollusks have also convergently evolved imply that the interlocking pattern emerges as the result of epi- a bivalved shell that displays an apparently mundane, yet strik- genetic interactions modulating the behavior of the secreting ing feature from a developmental point of view: When the shell mantle during shell development. is closed, the 2 valve edges meet each other in a commissure that Here, we provide a geometric and mechanical explanation forms a continuum with no gaps or overlaps despite the fact that for this morphological trait based on a detailed analysis of the each valve, secreted by 2 mantle lobes, may present antisymmet- shell geometry during growth and the physical interaction of the ric ornamental patterns of varying regularity and size. Interlock- shell-secreting soft mantle with both the rigid shell edge and ing is maintained throughout the entirety of development, even the opposing mantle lobe. -

Exputens) in Mexico, and a Review of All Species of This North American Subgenus
Natural History Museum /U, JH caY-^A 19*90 la Of Los Angeles County THE VELIGER © CMS, Inc., 1990 The Veliger 33(3):305-316 (July 2, 1990) First Occurrence of the Tethyan Bivalve Nayadina (.Exputens) in Mexico, and a Review of All Species of This North American Subgenus by RICHARD L. SQUIRES Department of Geological Sciences, California State University, Northridge, California 91330, USA Abstract. The malleid bivalve Nayadina (Exputens) has Old World Tethyan affinities but is known only from Eocene deposits in North America. Nayadina (Exputens) is reported for the first time from Mexico. About 50 specimens of N. (E.) batequensis sp. nov. were found in warm-water nearshore deposits of the middle lower Eocene part of the Bateque Formation, just south of Laguna San Ignacio, on the Pacific coast of Baja California Sur. The new species shows a wide range of morphologic variability especially where the beaks and auricles are located and how much they are developed. A review of the other species of Exputens, namely Nayadina (E.) llajasensis (Clark, 1934) from California and N. (E.) ocalensis (MacNeil, 1934) from Florida, Georgia, and North Carolina, revealed that they also have a wide range of morphologic variability. Nayadina (E.) alexi (Clark, 1934) is shown, herein, to be a junior synonym of N. (E.) llajasensis. The presence of a byssal sinus is recognized for the first time in Exputens. An epifaunal nestling mode of life, with attachment by byssus to hard substrate, can now be assumed for Exputens. INTRODUCTION species. It became necessary to thoroughly examine them, The macropaleontology of Eocene marine deposits in Baja and after such a study, it was found that the Bateque California Sur, Mexico, is largely an untouched subject. -

Freshwater Mussels of Maritime Canada: a Flashcard Guide
Freshwater Mussels of Maritime Canada: A Flashcard Guide In Wolastoqey, Mi’kmaw, French and English UMBO DORSAL MARGIN ANTERIOR POSTERIOR MARGIN MARGIN VENTRAL MARGIN Donald F. McAlpine, Mary C. Sollows, Jacqueline B. Madill and André L. Martel ISBN 978-0-919326-80-4 All photos copyright the Canadian Museum of Nature Acknowledgements: Funding for this publication provided by the Department of Fisheries and Oceans and the New Brunswick Museum. Special thanks to Ree Brennin Houston, Department of Fisheries and Oceans; Anne Hamilton, Brent Suttie, New Brunswick Archaeological Services Branch, and indigenous language translators Allan Tremblay (Wolastoqiyk), George Paul, Howard Augustine, and Karen Narvey (Mi’kmaw). Citation: McAlpine, D.F., M.C. Sollows, J. B. Madill, and A. L. Martel. 2018. Freshwater Mussels of Maritime Canada: A Flashcard Guide in Wolastoqey, Mi’kmaw, French and English. New Brunswick Museum, Saint John, New Brunswick, and Canadian Museum of Nature, Ottawa, Canada. Use in conjunction with Martel, A. L.,D.F. McAlpine, J. Madill, D. Sabine, A. Paquet, M. Pulsifer and M. Elderkin. 2010. Pp. 551-598. Freshwater Mussels (Bivalvia: Margaritiferidae, Unionidae) of the Atlantic Maritime Ecozone. In D.F. McAlpine and I.M. Smith (eds.). Assessment of Species Diversity in the Atlantic Maritime Ecozone. NRC Research Press, National Research Council of Canada, Ottawa, ON. 785 pp. Nedeau, E.J., M.A. McCollough, and B.I. Swartz. 2000. Freshwater Mussels of Maine. Maine Department of Inland Fisheries and Wildlife, Augusta, ME, 118 pp.