Shared and Species-Specific Patterns of Nascent Y Chromosome Evolution in Two Guppy Species
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

1 Gene Activation Precedes DNA Demethylation in Response to Infection in Human Dendritic Cells
bioRxiv preprint doi: https://doi.org/10.1101/358531; this version posted June 29, 2018. The copyright holder for this preprint (which was not certified by peer review) is the author/funder, who has granted bioRxiv a license to display the preprint in perpetuity. It is made available under aCC-BY-NC-ND 4.0 International license. 1 Gene activation precedes DNA demethylation in response to infection in human dendritic cells 2 3 Alain Pacis1,2‡, Florence Mailhot-Léonard1,2‡, Ludovic Tailleux3,4, Haley E Randolph1,2, Vania Yotova1, 4 Anne Dumaine1, Jean-Christophe Grenier1, Luis B Barreiro1,5,6*, 5 6 1CHU Sainte-Justine Research Center, Department of Genetics, Montreal, H3T1C5, Canada; 2University 7 of Montreal, Department of Biochemistry, Montreal, H3T1J4, Canada; 3Institut Pasteur, Mycobacterial 8 Genetics Unit, Paris, 75015, France; 4Institut Pasteur, Unit for Integrated Mycobacterial Pathogenomics, 9 CNRS UMR 3525, Paris, 75015, France; 5University of Montreal, Department of Pediatrics, Montreal, 10 H3T1J4, Canada; 6University of Chicago, Department of Medicine, Genetics Section, Chicago, Illinois, 11 USA. 12 13 14 ‡ These authors equally contributed to this study. 15 *Correspondence to: [email protected] 16 Running title: Epigenetic changes in response to infection 17 Keywords: DNA Methylation, Infection, Enhancers, Gene regulation 18 1 bioRxiv preprint doi: https://doi.org/10.1101/358531; this version posted June 29, 2018. The copyright holder for this preprint (which was not certified by peer review) is the author/funder, who has granted bioRxiv a license to display the preprint in perpetuity. It is made available under aCC-BY-NC-ND 4.0 International license. -
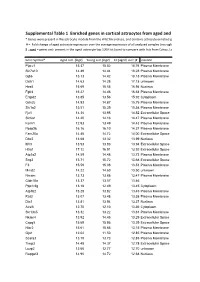
Supplemental Table 1 Enriched Genes in Cortical Astrocytes from Aged
Supplemental Table 1 Enriched genes in cortical astrocytes from aged and young-adult mice * Genes were present in the astrocyte module from the WGCNA analysis, and contains astrocyte enriched genes compared to microglia and oligodendrocytes # = Fold change of aged astrocyte expression over the average expression of all analyzed samples (microglia, astrocytes: young, old, with and without myelin contamination) $ ; aged = genes only present in the aged astrocyte top 1000 list (used to compare with lists from Cahoy, Lovatt, Doyle; see Fig. 4B), all = genes present in all astrocyte top 1000 lists Gene Symbol* Aged astr. (log2) Young astr.(log2) FC (aged/ aver.)# Location Ptprz1 15.37 15.02 18.76 Plasma Membrane Slc7a10 14.49 14.44 18.28 Plasma Membrane Gjb6 15.13 14.42 18.18 Plasma Membrane Dclk1 14.63 14.28 17.18 unknown Hes5 15.69 15.55 16.94 Nucleus Fgfr3 15.27 14.46 16.54 Plasma Membrane Entpd2 13.85 13.56 15.92 Cytoplasm Grin2c 14.93 14.87 15.75 Plasma Membrane Slc1a2 15.51 15.39 15.58 Plasma Membrane Fjx1 14.36 13.98 14.52 Extracellular Space Slc6a1 14.20 14.16 14.47 Plasma Membrane Kcnk1 12.93 13.49 14.43 Plasma Membrane Ppap2b 16.16 16.10 14.37 Plasma Membrane Fam20a 14.48 14.72 14.00 Extracellular Space Dbx2 13.68 13.32 13.99 Nucleus Itih3 13.93 13.93 13.94 Extracellular Space Htra1 17.12 16.91 13.92 Extracellular Space Atp1a2 14.59 14.48 13.73 Plasma Membrane Scg3 15.71 15.72 13.68 Extracellular Space F3 15.59 15.08 13.51 Plasma Membrane Mmd2 14.22 14.60 13.50 unknown Nrcam 13.73 13.88 13.47 Plasma Membrane Cldn10a 13.37 13.57 13.46 -

The Evolution of the Placenta Drives a Shift in Sexual Selection in Livebearing Fish
LETTER doi:10.1038/nature13451 The evolution of the placenta drives a shift in sexual selection in livebearing fish B. J. A. Pollux1,2, R. W. Meredith1,3, M. S. Springer1, T. Garland1 & D. N. Reznick1 The evolution of the placenta from a non-placental ancestor causes a species produce large, ‘costly’ (that is, fully provisioned) eggs5,6, gaining shift of maternal investment from pre- to post-fertilization, creating most reproductive benefits by carefully selecting suitable mates based a venue for parent–offspring conflicts during pregnancy1–4. Theory on phenotype or behaviour2. These females, however, run the risk of mat- predicts that the rise of these conflicts should drive a shift from a ing with genetically inferior (for example, closely related or dishonestly reliance on pre-copulatory female mate choice to polyandry in conjunc- signalling) males, because genetically incompatible males are generally tion with post-zygotic mechanisms of sexual selection2. This hypoth- not discernable at the phenotypic level10. Placental females may reduce esis has not yet been empirically tested. Here we apply comparative these risks by producing tiny, inexpensive eggs and creating large mixed- methods to test a key prediction of this hypothesis, which is that the paternity litters by mating with multiple males. They may then rely on evolution of placentation is associated with reduced pre-copulatory the expression of the paternal genomes to induce differential patterns of female mate choice. We exploit a unique quality of the livebearing fish post-zygotic maternal investment among the embryos and, in extreme family Poeciliidae: placentas have repeatedly evolved or been lost, cases, divert resources from genetically defective (incompatible) to viable creating diversity among closely related lineages in the presence or embryos1–4,6,11. -

Nuclear Organization and the Epigenetic Landscape of the Mus Musculus X-Chromosome Alicia Liu University of Connecticut - Storrs, [email protected]
University of Connecticut OpenCommons@UConn Doctoral Dissertations University of Connecticut Graduate School 8-9-2019 Nuclear Organization and the Epigenetic Landscape of the Mus musculus X-Chromosome Alicia Liu University of Connecticut - Storrs, [email protected] Follow this and additional works at: https://opencommons.uconn.edu/dissertations Recommended Citation Liu, Alicia, "Nuclear Organization and the Epigenetic Landscape of the Mus musculus X-Chromosome" (2019). Doctoral Dissertations. 2273. https://opencommons.uconn.edu/dissertations/2273 Nuclear Organization and the Epigenetic Landscape of the Mus musculus X-Chromosome Alicia J. Liu, Ph.D. University of Connecticut, 2019 ABSTRACT X-linked imprinted genes have been hypothesized to contribute parent-of-origin influences on social cognition. A cluster of imprinted genes Xlr3b, Xlr4b, and Xlr4c, implicated in cognitive defects, are maternally expressed and paternally silent in the murine brain. These genes defy classic mechanisms of autosomal imprinting, suggesting a novel method of imprinted gene regulation. Using Xlr3b and Xlr4c as bait, this study uses 4C-Seq on neonatal whole brain of a 39,XO mouse model, to provide the first in-depth analysis of chromatin dynamics surrounding an imprinted locus on the X-chromosome. Significant differences in long-range contacts exist be- tween XM and XP monosomic samples. In addition, XM interaction profiles contact a greater number of genes linked to cognitive impairment, abnormality of the nervous system, and abnormality of higher mental function. This is not a pattern that is unique to the imprinted Xlr3/4 locus. Additional Alicia J. Liu - University of Connecticut - 2019 4C-Seq experiments show that other genes on the X-chromosome, implicated in intellectual disability and/or ASD, also produce more maternal contacts to other X-linked genes linked to cognitive impairment. -

Environmental Influences on Endothelial Gene Expression
ENDOTHELIAL CELL GENE EXPRESSION John Matthew Jeff Herbert Supervisors: Prof. Roy Bicknell and Dr. Victoria Heath PhD thesis University of Birmingham August 2012 University of Birmingham Research Archive e-theses repository This unpublished thesis/dissertation is copyright of the author and/or third parties. The intellectual property rights of the author or third parties in respect of this work are as defined by The Copyright Designs and Patents Act 1988 or as modified by any successor legislation. Any use made of information contained in this thesis/dissertation must be in accordance with that legislation and must be properly acknowledged. Further distribution or reproduction in any format is prohibited without the permission of the copyright holder. ABSTRACT Tumour angiogenesis is a vital process in the pathology of tumour development and metastasis. Targeting markers of tumour endothelium provide a means of targeted destruction of a tumours oxygen and nutrient supply via destruction of tumour vasculature, which in turn ultimately leads to beneficial consequences to patients. Although current anti -angiogenic and vascular targeting strategies help patients, more potently in combination with chemo therapy, there is still a need for more tumour endothelial marker discoveries as current treatments have cardiovascular and other side effects. For the first time, the analyses of in-vivo biotinylation of an embryonic system is performed to obtain putative vascular targets. Also for the first time, deep sequencing is applied to freshly isolated tumour and normal endothelial cells from lung, colon and bladder tissues for the identification of pan-vascular-targets. Integration of the proteomic, deep sequencing, public cDNA libraries and microarrays, delivers 5,892 putative vascular targets to the science community. -

A Computational Approach for Defining a Signature of Β-Cell Golgi Stress in Diabetes Mellitus
Page 1 of 781 Diabetes A Computational Approach for Defining a Signature of β-Cell Golgi Stress in Diabetes Mellitus Robert N. Bone1,6,7, Olufunmilola Oyebamiji2, Sayali Talware2, Sharmila Selvaraj2, Preethi Krishnan3,6, Farooq Syed1,6,7, Huanmei Wu2, Carmella Evans-Molina 1,3,4,5,6,7,8* Departments of 1Pediatrics, 3Medicine, 4Anatomy, Cell Biology & Physiology, 5Biochemistry & Molecular Biology, the 6Center for Diabetes & Metabolic Diseases, and the 7Herman B. Wells Center for Pediatric Research, Indiana University School of Medicine, Indianapolis, IN 46202; 2Department of BioHealth Informatics, Indiana University-Purdue University Indianapolis, Indianapolis, IN, 46202; 8Roudebush VA Medical Center, Indianapolis, IN 46202. *Corresponding Author(s): Carmella Evans-Molina, MD, PhD ([email protected]) Indiana University School of Medicine, 635 Barnhill Drive, MS 2031A, Indianapolis, IN 46202, Telephone: (317) 274-4145, Fax (317) 274-4107 Running Title: Golgi Stress Response in Diabetes Word Count: 4358 Number of Figures: 6 Keywords: Golgi apparatus stress, Islets, β cell, Type 1 diabetes, Type 2 diabetes 1 Diabetes Publish Ahead of Print, published online August 20, 2020 Diabetes Page 2 of 781 ABSTRACT The Golgi apparatus (GA) is an important site of insulin processing and granule maturation, but whether GA organelle dysfunction and GA stress are present in the diabetic β-cell has not been tested. We utilized an informatics-based approach to develop a transcriptional signature of β-cell GA stress using existing RNA sequencing and microarray datasets generated using human islets from donors with diabetes and islets where type 1(T1D) and type 2 diabetes (T2D) had been modeled ex vivo. To narrow our results to GA-specific genes, we applied a filter set of 1,030 genes accepted as GA associated. -

Description of Poecilia (Acanthophacelus) Wingei N
Contributions to Zoology, 74 (1/2) 97-115 (2005) Description of Poecilia (Acanthophacelus) wingei n. sp. from the Paría Peninsula, Venezuela, including notes on Acanthophacelus Eigenmann, 1907 and other subgenera of Poecilia Bloch and Schneider, 1801 (Teleostei, Cyprinodontiformes, Poeciliidae) Fred. N. Poeser1, Michael Kempkes2, Isaäc J. H. Isbrücker1 1 Zoological Museum Amsterdam, University of Amsterdam, P.O. Box 94766, 1090 GT, Amsterdam, The Nether- lands, e-mail: [email protected]; 2 Am Mühlenberg 25, D-46419 Isselburg – Anholt, Germany Keywords: Guppies, Poecilia reticulata, P. wingei, melanophore patterns, character displacement, Paría Peninsula, despeciation Abstract Remarks on the 'Endler’s live-bearer' ...................................... 113 Acknowledgements ..................................................................... 114 The taxonomy of the common guppy, Poecilia reticulata Peters, References ..................................................................................... 114 1859, is reviewed and the closely related Campoma guppy, P. wingei n. sp., is described. Formerly, the common guppy was not judged to be closely related to any other species of Poecilia, but Introduction the new species is the second species to be allocated in the sub- genus Acanthophacelus Eigenmann, 1907. The recognition of P. wingei results from observed character displacement, i.e., on the The common guppy, Poecilia reticulata Peters, 1859, interaction between two closely related species in a shared envi- has a long history as -

Transcriptional Control of Tissue-Resident Memory T Cell Generation
Transcriptional control of tissue-resident memory T cell generation Filip Cvetkovski Submitted in partial fulfillment of the requirements for the degree of Doctor of Philosophy in the Graduate School of Arts and Sciences COLUMBIA UNIVERSITY 2019 © 2019 Filip Cvetkovski All rights reserved ABSTRACT Transcriptional control of tissue-resident memory T cell generation Filip Cvetkovski Tissue-resident memory T cells (TRM) are a non-circulating subset of memory that are maintained at sites of pathogen entry and mediate optimal protection against reinfection. Lung TRM can be generated in response to respiratory infection or vaccination, however, the molecular pathways involved in CD4+TRM establishment have not been defined. Here, we performed transcriptional profiling of influenza-specific lung CD4+TRM following influenza infection to identify pathways implicated in CD4+TRM generation and homeostasis. Lung CD4+TRM displayed a unique transcriptional profile distinct from spleen memory, including up-regulation of a gene network induced by the transcription factor IRF4, a known regulator of effector T cell differentiation. In addition, the gene expression profile of lung CD4+TRM was enriched in gene sets previously described in tissue-resident regulatory T cells. Up-regulation of immunomodulatory molecules such as CTLA-4, PD-1, and ICOS, suggested a potential regulatory role for CD4+TRM in tissues. Using loss-of-function genetic experiments in mice, we demonstrate that IRF4 is required for the generation of lung-localized pathogen-specific effector CD4+T cells during acute influenza infection. Influenza-specific IRF4−/− T cells failed to fully express CD44, and maintained high levels of CD62L compared to wild type, suggesting a defect in complete differentiation into lung-tropic effector T cells. -

Implication of M6a Mrna Methylation in Susceptibility to Inflammatory
epigenomes Article Implication of m6A mRNA Methylation in Susceptibility to Inflammatory Bowel Disease Maialen Sebastian-delaCruz 1,2 , Ane Olazagoitia-Garmendia 1,2 , Itziar Gonzalez-Moro 2,3 , Izortze Santin 2,3,4 , Koldo Garcia-Etxebarria 5 and Ainara Castellanos-Rubio 1,2,4,6,* 1 Department of Genetics, Physical Anthropology and Animal Fisiology, University of the Basque Country, 48940 Leioa, Spain; [email protected] (M.S.-d.); [email protected] (A.O.-G.) 2 Biocruces Bizkaia Health Research Institute, 48903 Barakaldo, Spain; [email protected] (I.G.-M.); [email protected] (I.S.) 3 Department of Biochemistry and Molecular Biology, University of the Basque Country, 48940 Leioa, Spain 4 CIBER (Centro de Investigación Biomédica en Red) de Diabetes y Enfermedades Metabólicas Asociadas (CIBERDEM), Instituto de Salud Carlos III, 28029 Madrid, Spain 5 Hepatic and Gastrointestinal Disease Area, IIS Biodonostia, 20014 Donostia, Spain; [email protected] 6 Ikerbasque, Basque Foundation for Science, 48013 Bilbao, Spain * Correspondence: [email protected] Received: 29 June 2020; Accepted: 28 July 2020; Published: 3 August 2020 Abstract: Inflammatory bowel disease (IBD) is a chronic inflammatory condition of the gastrointestinal tract that develops due to the interaction between genetic and environmental factors. More than 160 loci have been associated with IBD, but the functional implication of many of the associated genes remains unclear. N6-Methyladenosine (m6A) is the most abundant internal modification in mRNA. m6A methylation regulates many aspects of mRNA metabolism, playing important roles in the development of several pathologies. Interestingly, SNPs located near or within m6A motifs have been proposed as possible contributors to disease pathogenesis. -

Poecilia Wingei
MASARYKOVA UNIVERZITA PŘÍRODOVĚDECKÁ FAKULTA ÚSTAV BOTANIKY A ZOOLOGIE AKADEMIE VĚD ČR ÚSTAV BIOLOGIE OBRATLOVCŮ, V.V.I. Personality, reprodukční strategie a pohlavní výběr u vybraných taxonů ryb Disertační práce Radomil Řežucha ŠKOLITEL: doc. RNDr. MARTIN REICHARD, Ph.D. BRNO 2014 Bibliografický záznam Autor: Mgr. Radomil Řežucha Přírodovědecká fakulta, Masarykova univerzita Ústav botaniky a zoologie Název práce: Personality, reprodukční strategie a pohlavní výběr u vybraných taxonů ryb Studijní program: Biologie Studijní obor: Zoologie Školitel: doc. RNDr. Martin Reichard, Ph.D. Akademie věd ČR Ústav biologie obratlovců, v.v.i. Akademický rok: 2013/2014 Počet stran: 139 Klíčová slova: Pohlavní výběr, alternativní rozmnožovací takti- ky, osobnostní znaky, sociální prostředí, zkuše- nost, Rhodeus amarus, Poecilia wingei Bibliographic Entry Author: Mgr. Radomil Řežucha Faculty of Science, Masaryk University Department of Botany and Zoology Title of Dissertation: Personalities, reproductive tactics and sexual selection in fishes Degree Programme: Biology Field of Study: Zoology Supervisor doc. RNDr. Martin Reichard, Ph.D. Academy of Sciences of the Czech Republic Institute of Vertebrate Biology, v.v.i. Academic Year: 2013/2014 Number of pages: 139 Keywords: Sexual selection, alternative mating tactics, per- sonality traits, social environment, experience, Rhodeus amarus, Poecilia wingei Abstrakt Vliv osobnostních znaků na alternativní reprodukční taktiky (charakteris- tické typy reprodukčního chování) patří mezi zanedbávané oblasti studia po- hlavního výběru. Současně bývá opomíjen i vliv sociálního prostředí a zkuše- nosti na tyto taktiky, a studium schopnosti jedinců v průběhu námluv mas- kovat své morfologické nedostatky. Jako studovaný systém alternativních rozmnožovacích taktik byl zvolen v přírodě nejběžnější komplex – sneaker × guarder (courter) komplex, popisující teritoriální a neteritoriální role samců. -

A Flexible Microfluidic System for Single-Cell Transcriptome Profiling
www.nature.com/scientificreports OPEN A fexible microfuidic system for single‑cell transcriptome profling elucidates phased transcriptional regulators of cell cycle Karen Davey1,7, Daniel Wong2,7, Filip Konopacki2, Eugene Kwa1, Tony Ly3, Heike Fiegler2 & Christopher R. Sibley 1,4,5,6* Single cell transcriptome profling has emerged as a breakthrough technology for the high‑resolution understanding of complex cellular systems. Here we report a fexible, cost‑efective and user‑ friendly droplet‑based microfuidics system, called the Nadia Instrument, that can allow 3′ mRNA capture of ~ 50,000 single cells or individual nuclei in a single run. The precise pressure‑based system demonstrates highly reproducible droplet size, low doublet rates and high mRNA capture efciencies that compare favorably in the feld. Moreover, when combined with the Nadia Innovate, the system can be transformed into an adaptable setup that enables use of diferent bufers and barcoded bead confgurations to facilitate diverse applications. Finally, by 3′ mRNA profling asynchronous human and mouse cells at diferent phases of the cell cycle, we demonstrate the system’s ability to readily distinguish distinct cell populations and infer underlying transcriptional regulatory networks. Notably this provided supportive evidence for multiple transcription factors that had little or no known link to the cell cycle (e.g. DRAP1, ZKSCAN1 and CEBPZ). In summary, the Nadia platform represents a promising and fexible technology for future transcriptomic studies, and other related applications, at cell resolution. Single cell transcriptome profling has recently emerged as a breakthrough technology for understanding how cellular heterogeneity contributes to complex biological systems. Indeed, cultured cells, microorganisms, biopsies, blood and other tissues can be rapidly profled for quantifcation of gene expression at cell resolution. -

Supplementary File 2A Revised
Supplementary file 2A. Differentially expressed genes in aldosteronomas compared to all other samples, ranked according to statistical significance. Missing values were not allowed in aldosteronomas, but to a maximum of five in the other samples. Acc UGCluster Name Symbol log Fold Change P - Value Adj. P-Value B R99527 Hs.8162 Hypothetical protein MGC39372 MGC39372 2,17 6,3E-09 5,1E-05 10,2 AA398335 Hs.10414 Kelch domain containing 8A KLHDC8A 2,26 1,2E-08 5,1E-05 9,56 AA441933 Hs.519075 Leiomodin 1 (smooth muscle) LMOD1 2,33 1,3E-08 5,1E-05 9,54 AA630120 Hs.78781 Vascular endothelial growth factor B VEGFB 1,24 1,1E-07 2,9E-04 7,59 R07846 Data not found 3,71 1,2E-07 2,9E-04 7,49 W92795 Hs.434386 Hypothetical protein LOC201229 LOC201229 1,55 2,0E-07 4,0E-04 7,03 AA454564 Hs.323396 Family with sequence similarity 54, member B FAM54B 1,25 3,0E-07 5,2E-04 6,65 AA775249 Hs.513633 G protein-coupled receptor 56 GPR56 -1,63 4,3E-07 6,4E-04 6,33 AA012822 Hs.713814 Oxysterol bining protein OSBP 1,35 5,3E-07 7,1E-04 6,14 R45592 Hs.655271 Regulating synaptic membrane exocytosis 2 RIMS2 2,51 5,9E-07 7,1E-04 6,04 AA282936 Hs.240 M-phase phosphoprotein 1 MPHOSPH -1,40 8,1E-07 8,9E-04 5,74 N34945 Hs.234898 Acetyl-Coenzyme A carboxylase beta ACACB 0,87 9,7E-07 9,8E-04 5,58 R07322 Hs.464137 Acyl-Coenzyme A oxidase 1, palmitoyl ACOX1 0,82 1,3E-06 1,2E-03 5,35 R77144 Hs.488835 Transmembrane protein 120A TMEM120A 1,55 1,7E-06 1,4E-03 5,07 H68542 Hs.420009 Transcribed locus 1,07 1,7E-06 1,4E-03 5,06 AA410184 Hs.696454 PBX/knotted 1 homeobox 2 PKNOX2 1,78 2,0E-06