Molecular Genetics of Relapsed Diffuse Large B-Cell
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

WO 2019/079361 Al 25 April 2019 (25.04.2019) W 1P O PCT
(12) INTERNATIONAL APPLICATION PUBLISHED UNDER THE PATENT COOPERATION TREATY (PCT) (19) World Intellectual Property Organization I International Bureau (10) International Publication Number (43) International Publication Date WO 2019/079361 Al 25 April 2019 (25.04.2019) W 1P O PCT (51) International Patent Classification: CA, CH, CL, CN, CO, CR, CU, CZ, DE, DJ, DK, DM, DO, C12Q 1/68 (2018.01) A61P 31/18 (2006.01) DZ, EC, EE, EG, ES, FI, GB, GD, GE, GH, GM, GT, HN, C12Q 1/70 (2006.01) HR, HU, ID, IL, IN, IR, IS, JO, JP, KE, KG, KH, KN, KP, KR, KW, KZ, LA, LC, LK, LR, LS, LU, LY, MA, MD, ME, (21) International Application Number: MG, MK, MN, MW, MX, MY, MZ, NA, NG, NI, NO, NZ, PCT/US2018/056167 OM, PA, PE, PG, PH, PL, PT, QA, RO, RS, RU, RW, SA, (22) International Filing Date: SC, SD, SE, SG, SK, SL, SM, ST, SV, SY, TH, TJ, TM, TN, 16 October 2018 (16. 10.2018) TR, TT, TZ, UA, UG, US, UZ, VC, VN, ZA, ZM, ZW. (25) Filing Language: English (84) Designated States (unless otherwise indicated, for every kind of regional protection available): ARIPO (BW, GH, (26) Publication Language: English GM, KE, LR, LS, MW, MZ, NA, RW, SD, SL, ST, SZ, TZ, (30) Priority Data: UG, ZM, ZW), Eurasian (AM, AZ, BY, KG, KZ, RU, TJ, 62/573,025 16 October 2017 (16. 10.2017) US TM), European (AL, AT, BE, BG, CH, CY, CZ, DE, DK, EE, ES, FI, FR, GB, GR, HR, HU, ΓΕ , IS, IT, LT, LU, LV, (71) Applicant: MASSACHUSETTS INSTITUTE OF MC, MK, MT, NL, NO, PL, PT, RO, RS, SE, SI, SK, SM, TECHNOLOGY [US/US]; 77 Massachusetts Avenue, TR), OAPI (BF, BJ, CF, CG, CI, CM, GA, GN, GQ, GW, Cambridge, Massachusetts 02139 (US). -

Supplementary Table S2
1-high in cerebrotropic Gene P-value patients Definition BCHE 2.00E-04 1 Butyrylcholinesterase PLCB2 2.00E-04 -1 Phospholipase C, beta 2 SF3B1 2.00E-04 -1 Splicing factor 3b, subunit 1 BCHE 0.00022 1 Butyrylcholinesterase ZNF721 0.00028 -1 Zinc finger protein 721 GNAI1 0.00044 1 Guanine nucleotide binding protein (G protein), alpha inhibiting activity polypeptide 1 GNAI1 0.00049 1 Guanine nucleotide binding protein (G protein), alpha inhibiting activity polypeptide 1 PDE1B 0.00069 -1 Phosphodiesterase 1B, calmodulin-dependent MCOLN2 0.00085 -1 Mucolipin 2 PGCP 0.00116 1 Plasma glutamate carboxypeptidase TMX4 0.00116 1 Thioredoxin-related transmembrane protein 4 C10orf11 0.00142 1 Chromosome 10 open reading frame 11 TRIM14 0.00156 -1 Tripartite motif-containing 14 APOBEC3D 0.00173 -1 Apolipoprotein B mRNA editing enzyme, catalytic polypeptide-like 3D ANXA6 0.00185 -1 Annexin A6 NOS3 0.00209 -1 Nitric oxide synthase 3 SELI 0.00209 -1 Selenoprotein I NYNRIN 0.0023 -1 NYN domain and retroviral integrase containing ANKFY1 0.00253 -1 Ankyrin repeat and FYVE domain containing 1 APOBEC3F 0.00278 -1 Apolipoprotein B mRNA editing enzyme, catalytic polypeptide-like 3F EBI2 0.00278 -1 Epstein-Barr virus induced gene 2 ETHE1 0.00278 1 Ethylmalonic encephalopathy 1 PDE7A 0.00278 -1 Phosphodiesterase 7A HLA-DOA 0.00305 -1 Major histocompatibility complex, class II, DO alpha SOX13 0.00305 1 SRY (sex determining region Y)-box 13 ABHD2 3.34E-03 1 Abhydrolase domain containing 2 MOCS2 0.00334 1 Molybdenum cofactor synthesis 2 TTLL6 0.00365 -1 Tubulin tyrosine ligase-like family, member 6 SHANK3 0.00394 -1 SH3 and multiple ankyrin repeat domains 3 ADCY4 0.004 -1 Adenylate cyclase 4 CD3D 0.004 -1 CD3d molecule, delta (CD3-TCR complex) (CD3D), transcript variant 1, mRNA. -

Anti-HIST1H1E K51ac Antibody
FOR RESEARCH USE ONLY! 02/20 Anti-HIST1H1E K51ac Antibody CATALOG NO.: A2051-100 (100 µl) BACKGROUND DESCRIPTION: Histones are basic nuclear proteins responsible for nucleosome structure of the chromosomal fiber in eukaryotes. Two molecules of each of the four core histones (H2A, H2B, H3, and H4) form an octamer, around which approximately 146 bp of DNA is wrapped in repeating units, called nucleosomes. The linker histone, H1, interacts with linker DNA between nucleosomes and functions in the compaction of chromatin into higher order structures. This gene is intronless and encodes a replication-dependent histone that is a member of the histone H1 family. Transcripts from this gene lack poly A tails but instead contain a palindromic termination element. This gene is found in the large histone gene cluster on chromosome 6. Histone H1.4 (Histone H1b) (Histone H1s-4), HIST1H1E, H1F4 ALTERNATE NAMES: ANTIBODY TYPE: Polyclonal HOST/ISOTYPE: Rabbit / IgG IMMUNOGEN: Acetylated peptide sequence targeting residues around Lysine 51 of human Histone H1.4 PURIFICATION: Antigen Affinity purified FORM: Liquid FORMULATION: In 0.01 M PBS, pH 7.4, 50% Glycerol, 0.03% proclin 300 SPECIES REACTIVITY: Human STORAGE CONDITIONS: Store at -20ºC. Avoid freeze / thaw cycles. APPLICATIONS AND USAGE: ICC 1:20-1:200, IF 1:50-1:200 This information is only intended as a guide. The optimal dilutions must be determined by the user Immunocytochemistry analysis of HeLa cells using Anti-HIST1H1E K51ac antibody at dilution of 1:100. Immunofluorescent analysis of HeLa cells (treated with sodium butyrate, 30 mM, 4 hrs) using Anti-HIST1H1E K51ac antibody at dilution of 1:100 and Alexa Fluor 488-conjugated Goat Anti-Rabbit IgG (H+L) as secondary antibody. -

Genomic Analyses of PMBL Reveal New Drivers and Mechanisms of Sensitivity to PD-1 Blockade
Regular Article LYMPHOID NEOPLASIA Genomic analyses of PMBL reveal new drivers and mechanisms of sensitivity to PD-1 blockade Bjoern Chapuy,1,2,* Chip Stewart,3,* Andrew J. Dunford,3,* Jaegil Kim,3 Kirsty Wienand,1 Atanas Kamburov,3 Gabriel K. Griffin,4 Pei-Hsuan Chen,4 Ana Lako,4 Robert A. Redd,5 Claire M. Cote,1 Matthew D. Ducar,6 Aaron R. Thorner,6 Scott J. Rodig,4 Gad Getz,3,7,8,† Downloaded from https://ashpublications.org/blood/article-pdf/134/26/2369/1549702/bloodbld2019002067.pdf by Margaret Shipp on 30 December 2019 and Margaret A. Shipp1,† 1Department of Medical Oncology, Dana-Farber Cancer Institute, Harvard Medical School, Boston, MA; 2Department of Hematology and Oncology, University Medical Center Gottingen,¨ Gottingen,¨ Germany; 3Broad Institute of Harvard and Massachusetts Institute of Technology, Cambridge, MA; 4Department of Pathology, Brigham and Women’s Hospital, Harvard Medical School, Boston, MA; 5Department of Data Sciences and 6Center for Cancer Genome Discovery, Dana-Farber Cancer Institute, Boston, MA; and 7Department of Pathology and 8Center for Cancer Research, Massachusetts General Hospital, Harvard Medical School, Boston, MA KEY POINTS Primary mediastinal large B-cell lymphomas (PMBLs) are aggressive tumors that typically present as large mediastinal masses in young women. PMBLs share clinical, transcriptional, l Comprehensive genomic analyses of and molecular features with classical Hodgkin lymphoma (cHL), including constitutive PMBL reveal new activation of nuclear factor kB (NF-kB), JAK/STAT signaling, and programmed cell death genetic drivers such protein 1 (PD-1)–mediated immune evasion. The demonstrated efficacy of PD-1 blockade in as ZNF217. relapsed/refractory PMBLs led to recent approval by the US Food and Drug Administration l High mutational and underscored the importance of characterizing targetable genetic vulnerabilities in this burden, MSI, and disease. -

Transcriptome Analysis of Human Diabetic Kidney Disease
ORIGINAL ARTICLE Transcriptome Analysis of Human Diabetic Kidney Disease Karolina I. Woroniecka,1 Ae Seo Deok Park,1 Davoud Mohtat,2 David B. Thomas,3 James M. Pullman,4 and Katalin Susztak1,5 OBJECTIVE—Diabetic kidney disease (DKD) is the single cases, mild and then moderate mesangial expansion can be leading cause of kidney failure in the U.S., for which a cure has observed. In general, diabetic kidney disease (DKD) is not yet been found. The aim of our study was to provide an considered a nonimmune-mediated degenerative disease unbiased catalog of gene-expression changes in human diabetic of the glomerulus; however, it has long been noted that kidney biopsy samples. complement and immunoglobulins sometimes can be de- — tected in diseased glomeruli, although their role and sig- RESEARCH DESIGN AND METHODS Affymetrix expression fi arrays were used to identify differentially regulated transcripts in ni cance is not clear (4). 44 microdissected human kidney samples. The DKD samples were The understanding of DKD has been challenged by multi- significant for their racial diversity and decreased glomerular ple issues. First, the diagnosis of DKD usually is made using filtration rate (~20–30 mL/min). Stringent statistical analysis, using clinical criteria, and kidney biopsy often is not performed. the Benjamini-Hochberg corrected two-tailed t test, was used to According to current clinical practice, the development of identify differentially expressed transcripts in control and diseased albuminuria in patients with diabetes is sufficient to make the glomeruli and tubuli. Two different Web-based algorithms were fi diagnosis of DKD (5). We do not understand the correlation used to de ne differentially regulated pathways. -

Literature Mining Sustains and Enhances Knowledge Discovery from Omic Studies
LITERATURE MINING SUSTAINS AND ENHANCES KNOWLEDGE DISCOVERY FROM OMIC STUDIES by Rick Matthew Jordan B.S. Biology, University of Pittsburgh, 1996 M.S. Molecular Biology/Biotechnology, East Carolina University, 2001 M.S. Biomedical Informatics, University of Pittsburgh, 2005 Submitted to the Graduate Faculty of School of Medicine in partial fulfillment of the requirements for the degree of Doctor of Philosophy University of Pittsburgh 2016 UNIVERSITY OF PITTSBURGH SCHOOL OF MEDICINE This dissertation was presented by Rick Matthew Jordan It was defended on December 2, 2015 and approved by Shyam Visweswaran, M.D., Ph.D., Associate Professor Rebecca Jacobson, M.D., M.S., Professor Songjian Lu, Ph.D., Assistant Professor Dissertation Advisor: Vanathi Gopalakrishnan, Ph.D., Associate Professor ii Copyright © by Rick Matthew Jordan 2016 iii LITERATURE MINING SUSTAINS AND ENHANCES KNOWLEDGE DISCOVERY FROM OMIC STUDIES Rick Matthew Jordan, M.S. University of Pittsburgh, 2016 Genomic, proteomic and other experimentally generated data from studies of biological systems aiming to discover disease biomarkers are currently analyzed without sufficient supporting evidence from the literature due to complexities associated with automated processing. Extracting prior knowledge about markers associated with biological sample types and disease states from the literature is tedious, and little research has been performed to understand how to use this knowledge to inform the generation of classification models from ‘omic’ data. Using pathway analysis methods to better understand the underlying biology of complex diseases such as breast and lung cancers is state-of-the-art. However, the problem of how to combine literature- mining evidence with pathway analysis evidence is an open problem in biomedical informatics research. -
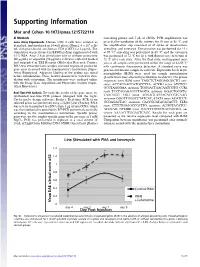
Supporting Information
Supporting Information Mor and Cohen 10.1073/pnas.1215722110 SI Methods containing primer and 5 μL of cDNA. PCR amplification was Gene Array Experiments. Human CD4 T cells were isolated as preceded by incubation of the mixture for 10 min at 95 °C, and described, and incubated in 24-well plates (Nunc), 4 × 106 cells/ the amplification step consisted of 45 cycles of denaturation, mL with plate-bound anti-human CD3 (OKT3) at 2 μg/mL. The annealing, and extension. Denaturation was performed for 15 s stimulation was performed in RPMI medium supplemented with at 95 °C; annealing was performed in 60 °C; and the extension 0.1% BSA. After 2 h of stimulation with or without cefuroxime was performed at 72 °C for 20 s, with fluorescence detection at (50 μg/mL) or ampicillin (50 μg/mL), cells were collected washed 72 °C after each cycle. After the final cycle, melting-point anal- and suspended in TRI Reagent (Molecular Research Center). yses of all samples were performed within the range of 62–95 °C RNA was extracted from samples and used to prepare probes for with continuous fluorescence detection. A standard curve was gene array in accord with the manufacturer’s instructions (Super- generated from one sample in each run. Expression levels of β2- Array Bioscience). Adequate labeling of the probes was tested microglobulin (B2M) were used for sample normalization before hybridization. Three healthy donors were tested in stim- (β-actin levels were affected by cefuroxime treatment). The primer ulation with cefuroxime. The membranes were analyzed online sequences were B2M sense TAGCTCTAGGAGGGCTG anti- with the Image Data Acquisition and Expression Analysis (Super- sense ACCACAACCATGCCTTA; ACVR2 sense ATCTCC- Array Bioscience). -

Genome-Wide Identification of Genes Regulating DNA Methylation Using Genetic Anchors for Causal Inference
bioRxiv preprint doi: https://doi.org/10.1101/823807; this version posted October 30, 2019. The copyright holder for this preprint (which was not certified by peer review) is the author/funder, who has granted bioRxiv a license to display the preprint in perpetuity. It is made available under aCC-BY 4.0 International license. Genome-wide identification of genes regulating DNA methylation using genetic anchors for causal inference Paul J. Hop1,2, René Luijk1, Lucia Daxinger3, Maarten van Iterson1, Koen F. Dekkers1, Rick Jansen4, BIOS Consortium5, Joyce B.J. van Meurs6, Peter A.C. ’t Hoen 7, M. Arfan Ikram8, Marleen M.J. van Greevenbroek9,10, Dorret I. Boomsma11, P. Eline Slagboom1, Jan H. Veldink2, Erik W. van Zwet12, Bastiaan T. Heijmans1* 1 Molecular Epidemiology, Department of Biomedical Data Sciences, Leiden University Medical Center, Leiden, 2333 ZC, The Netherlands 2 Department of Neurology, UMC Utrecht Brain Center, University Medical Centre Utrecht, Utrecht University, Utrecht 3584 CG, Netherlands 3 Department of Human Genetics, Leiden University Medical Center, Leiden, 2333 ZC, The Netherlands 4 Department of Psychiatry, Amsterdam UMC, Vrije Universiteit Amsterdam, Amsterdam Neuroscience, Amsterdam, 1081 HV, The Netherlands 5 Biobank-based Integrated Omics Study Consortium. For a complete list of authors, see the acknowledgements. 6 Department of Internal Medicine, Erasmus Medical Centre, Rotterdam, The Netherlands. 7 Centre for Molecular and Biomolecular Informatics, Radboud Institute for Molecular Life Sciences, Radboud University -

Deletions of IKZF1 and SPRED1 Are Associated with Poor Prognosis in A
Leukemia (2014) 28, 302–310 & 2014 Macmillan Publishers Limited All rights reserved 0887-6924/14 www.nature.com/leu ORIGINAL ARTICLE Deletions of IKZF1 and SPRED1 are associated with poor prognosis in a population-based series of pediatric B-cell precursor acute lymphoblastic leukemia diagnosed between 1992 and 2011 L Olsson1, A Castor2, M Behrendtz3, A Biloglav1, E Forestier4, K Paulsson1 and B Johansson1,5 Despite the favorable prognosis of childhood acute lymphoblastic leukemia (ALL), a substantial subset of patients relapses. As this occurs not only in the high risk but also in the standard/intermediate groups, the presently used risk stratification is suboptimal. The underlying mechanisms for treatment failure include the presence of genetic changes causing insensitivity to the therapy administered. To identify relapse-associated aberrations, we performed single-nucleotide polymorphism array analyses of 307 uniformly treated, consecutive pediatric ALL cases accrued during 1992–2011. Recurrent aberrations of 14 genes in patients who subsequently relapsed or had induction failure were detected. Of these, deletions/uniparental isodisomies of ADD3, ATP10A, EBF1, IKZF1, PAN3, RAG1, SPRED1 and TBL1XR1 were significantly more common in B-cell precursor ALL patients who relapsed compared with those remaining in complete remission. In univariate analyses, age (X10 years), white blood cell counts (4100 Â 109/l), t(9;22)(q34;q11), MLL rearrangements, near-haploidy and deletions of ATP10A, IKZF1, SPRED1 and the pseudoautosomal 1 regions on Xp/Yp were significantly associated with decreased 10-year event-free survival, with IKZF1 abnormalities being an independent risk factor in multivariate analysis irrespective of the risk group. Older age and deletions of IKZF1 and SPRED1 were also associated with poor overall survival. -

The Role of the NFAT Signalling Pathway in Diffuse Large B-Cell Lymphoma
The role of the NFAT signalling pathway in Diffuse Large B-cell Lymphoma Holly White Doctor of Philosophy Thesis September 2015 Supervisors: Dr Chris Bacon, Professor Neil Perkins & Dr Vikki Rand Northern Institute for Cancer Research Word count: 66,024 1 Abstract Diffuse Large B-Cell Lymphomas (DLBCL) are common, aggressive malignancies of mature B-lymphocytes that represent ~40% of lymphomas. Despite the widespread use of combined immunochemotherapy, approximately 50% of patients with DLBCL die from their disease. The two main DLBCL subgroups resemble activated B cells (ABC) or germinal centre B cells (GCB), where patients with ABC-DLBCL have significantly worse outcome. There is urgent need for novel therapeutic strategies in the treatment of DLBCL, which requires a better understanding of the molecular pathways upon which tumours depend. Accumulating evidence suggests that the signalling networks promoting and sustaining DLBCL derive from dysregulation of the normal pathways controlling B-lymphocyte activation and differentiation. There is increasing evidence indicating important roles for the NFAT family of transcription factors in DLBCL. Constitutively-active nuclear NFAT2 has been demonstrated in approximately 40% of primary DLBCL samples and NFAT has been shown to regulate a small number of genes associated with DLBCL growth/survival. This project investigated the role of NFAT in DLBCL. Nuclear localisation and activation of NFAT family members were characterised in a panel of DLBCL cell lines and chemical inhibition of calcineurin/NFAT, using Cyclosporin A (CsA), indicated dependency on the calcineurin/NFAT pathway for survival. Gene expression microarray analysis performed in DLBCL cell lines treated with CsA revealed potential NFAT target genes involved in the tumour microenvironment and anergy. -

Gene Ontology Functional Annotations and Pleiotropy
Network based analysis of genetic disease associations Sarah Gilman Submitted in partial fulfillment of the requirements for the degree of Doctor of Philosophy under the Executive Committee of the Graduate School of Arts and Sciences COLUMBIA UNIVERSITY 2014 © 2013 Sarah Gilman All Rights Reserved ABSTRACT Network based analysis of genetic disease associations Sarah Gilman Despite extensive efforts and many promising early findings, genome-wide association studies have explained only a small fraction of the genetic factors contributing to common human diseases. There are many theories about where this “missing heritability” might lie, but increasingly the prevailing view is that common variants, the target of GWAS, are not solely responsible for susceptibility to common diseases and a substantial portion of human disease risk will be found among rare variants. Relatively new, such variants have not been subject to purifying selection, and therefore may be particularly pertinent for neuropsychiatric disorders and other diseases with greatly reduced fecundity. Recently, several researchers have made great progress towards uncovering the genetics behind autism and schizophrenia. By sequencing families, they have found hundreds of de novo variants occurring only in affected individuals, both large structural copy number variants and single nucleotide variants. Despite studying large cohorts there has been little recurrence among the genes implicated suggesting that many hundreds of genes may underlie these complex phenotypes. The question -

Systematic Analysis and Biomarker Study for Alzheimer's Disease
Xinzhong Li, Haiyan Wang, Jintao Long, Genhua Pan, Taigang He, Oleg Anichtchi k, Robert Belshaw, Diego Albani , Paul Edison, Elaine K Green and James Scott Systematic analysis and biomarker study for Alzheimer’s disease Article (Published version) (Refereed) Original citation: Li, Xinzhong and Wang, Haiyan and Long, Jintao and Pan, Genhua and He, Taigang and Anichtchik, Oleg and Belshaw, Robert and Albani, Diego and Edison, Paul and Green, Elaine K. and Scott, James (2018) Systematic analysis and biomarker study for Alzheimer’s disease. Scientific Reports , 8 (17394). ISSN 2045-2322 DOI: https://doi.org/10.1038/s41598-018-35789-3 © 2018 Springer Nature Publishing AG CC -BY 4.0 This version available at: http://eprints.lse.ac.uk/id/eprint/91279 Available in LSE Research Online: December 2018 LSE has developed LSE Research Online so that users may access research output of the School. Copyright © and Moral Rights for the papers on this site are retained by the individual authors and/or other copyright owners. Users may download and/or print one copy of any article(s) in LSE Research Online to facilitate their private study or for non-commercial research. You may not engage in further distribution of the material or use it for any profit-making activities or any commercial gain. You may freely distribute the URL ( http://eprints.lse.ac.uk ) of the LSE Research Online website. ǤǤ Ȁ Systematic Analysis and Biomarker Study for Alzheimer’s Disease Xinzhong Li ͷ, Haiyan Wang, Jintao Longͷ, Genhua Pan, Taigang He ͺ, Oleg Anichtchikͷ, Robert Belshawͷ, Diego Albani ͻ, Paul Edisonͼ, Elaine K Greenͷ & James Scottͼ Received: 3 April 2018 Revealing the relationship between dysfunctional genes in blood and brain tissues from patients with Accepted: 28 October 2018 Alzheimer’s Disease (AD) will help us to understand the pathology of this disease.