UNIVERSITY of CALIFORNIA Los Angeles Dissecting the Regulatory
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Gprc5b Modulates Inflammatory Response in Glomerular Diseases
BASIC RESEARCH www.jasn.org GPRC5b Modulates Inflammatory Response in Glomerular Diseases via NF-kB Pathway Sonia Zambrano,1 Katja Möller-Hackbarth,1 Xidan Li,1 Patricia Q. Rodriguez,1 Emmanuelle Charrin,1 Angelina Schwarz,1 Jenny Nyström,2 Annika Östman Wernerson,3 Mark Lal,4 and Jaakko Patrakka1 1Karolinska Insitutet/AstraZeneca Integrated Cardio Metabolic Center, Department of Laboratory Medicine, Karolinska Institutet at Karolinska University Hospital Huddinge, Stockholm, Sweden; 2Department of Physiology, Institute of Neuroscience and Physiology, University of Gothenburg, Gothenburg, Sweden; 3Division of Renal Medicine, Department of Clinical Sciences, Intervention and Technology, Karolinska Institutet, Stockholm, Sweden; and 4Division of Bioscience, Department of Cardiovascular, Renal and Metabolic Diseases, Innovative Medicines Biotech Unit, AstraZeneca, Gothenburg, Sweden ABSTRACT Background Inflammatory processes play an important role in the pathogenesis of glomerulopathies. Finding novel ways to suppress glomerular inflammation may offer a new way to stop disease progression. However, the molecular mechanisms that initiate and drive inflammation in the glomerulus are still poorly understood. Methods We performed large-scale gene expression profiling of glomerulus-associated G protein– coupled receptors (GPCRs) to identify new potential therapeutic targets for glomerulopathies. The expression of Gprc5b in disease was analyzed using quantitative PCR and immunofluorescence, and by analyzing published microarray data sets. In vivo studies were carried out in a podocyte-specificGprc5b knockout mouse line. Mechanistic studies were performed in cultured human podocytes. Results We identified an orphan GPCR, Gprc5b, as a novel gene highly enriched in podocytes that was significantly upregulated in common human glomerulopathies, including diabetic nephropathy, IgA ne- phropathy, and lupus nephritis. Similar upregulation of Gprc5b was detected in LPS-induced nephropathy in mice. -

Transcriptional Control of Tissue-Resident Memory T Cell Generation
Transcriptional control of tissue-resident memory T cell generation Filip Cvetkovski Submitted in partial fulfillment of the requirements for the degree of Doctor of Philosophy in the Graduate School of Arts and Sciences COLUMBIA UNIVERSITY 2019 © 2019 Filip Cvetkovski All rights reserved ABSTRACT Transcriptional control of tissue-resident memory T cell generation Filip Cvetkovski Tissue-resident memory T cells (TRM) are a non-circulating subset of memory that are maintained at sites of pathogen entry and mediate optimal protection against reinfection. Lung TRM can be generated in response to respiratory infection or vaccination, however, the molecular pathways involved in CD4+TRM establishment have not been defined. Here, we performed transcriptional profiling of influenza-specific lung CD4+TRM following influenza infection to identify pathways implicated in CD4+TRM generation and homeostasis. Lung CD4+TRM displayed a unique transcriptional profile distinct from spleen memory, including up-regulation of a gene network induced by the transcription factor IRF4, a known regulator of effector T cell differentiation. In addition, the gene expression profile of lung CD4+TRM was enriched in gene sets previously described in tissue-resident regulatory T cells. Up-regulation of immunomodulatory molecules such as CTLA-4, PD-1, and ICOS, suggested a potential regulatory role for CD4+TRM in tissues. Using loss-of-function genetic experiments in mice, we demonstrate that IRF4 is required for the generation of lung-localized pathogen-specific effector CD4+T cells during acute influenza infection. Influenza-specific IRF4−/− T cells failed to fully express CD44, and maintained high levels of CD62L compared to wild type, suggesting a defect in complete differentiation into lung-tropic effector T cells. -

Genome-Wide Sirna Screen for Mediators of NF-Κb Activation
Genome-wide siRNA screen for mediators SEE COMMENTARY of NF-κB activation Benjamin E. Gewurza, Fadi Towficb,c,1, Jessica C. Marb,d,1, Nicholas P. Shinnersa,1, Kaoru Takasakia, Bo Zhaoa, Ellen D. Cahir-McFarlanda, John Quackenbushe, Ramnik J. Xavierb,c, and Elliott Kieffa,2 aDepartment of Medicine and Microbiology and Molecular Genetics, Channing Laboratory, Brigham and Women’s Hospital and Harvard Medical School, Boston, MA 02115; bCenter for Computational and Integrative Biology, Massachusetts General Hospital, Harvard Medical School, Boston, MA 02114; cProgram in Medical and Population Genetics, The Broad Institute of Massachusetts Institute of Technology and Harvard, Cambridge, MA 02142; dDepartment of Biostatistics, Harvard School of Public Health, Boston, MA 02115; and eDepartment of Biostatistics and Computational Biology and Department of Cancer Biology, Dana-Farber Cancer Institute, Boston, MA 02115 Contributed by Elliott Kieff, December 16, 2011 (sent for review October 2, 2011) Although canonical NFκB is frequently critical for cell proliferation, (RIPK1). TRADD engages TNFR-associated factor 2 (TRAF2), survival, or differentiation, NFκB hyperactivation can cause malig- which recruits the ubiquitin (Ub) E2 ligase UBC5 and the E3 nant, inflammatory, or autoimmune disorders. Despite intensive ligases cIAP1 and cIAP2. CIAP1/2 polyubiquitinate RIPK1 and study, mammalian NFκB pathway loss-of-function RNAi analyses TRAF2, which recruit and activate the K63-Ub binding proteins have been limited to specific protein classes. We therefore under- TAB1, TAB2, and TAB3, as well as their associated kinase took a human genome-wide siRNA screen for novel NFκB activa- MAP3K7 (TAK1). TAK1 in turn phosphorylates IKKβ activa- tion pathway components. Using an Epstein Barr virus latent tion loop serines to promote IKK activity (4). -

Supplementary Table S4. FGA Co-Expressed Gene List in LUAD
Supplementary Table S4. FGA co-expressed gene list in LUAD tumors Symbol R Locus Description FGG 0.919 4q28 fibrinogen gamma chain FGL1 0.635 8p22 fibrinogen-like 1 SLC7A2 0.536 8p22 solute carrier family 7 (cationic amino acid transporter, y+ system), member 2 DUSP4 0.521 8p12-p11 dual specificity phosphatase 4 HAL 0.51 12q22-q24.1histidine ammonia-lyase PDE4D 0.499 5q12 phosphodiesterase 4D, cAMP-specific FURIN 0.497 15q26.1 furin (paired basic amino acid cleaving enzyme) CPS1 0.49 2q35 carbamoyl-phosphate synthase 1, mitochondrial TESC 0.478 12q24.22 tescalcin INHA 0.465 2q35 inhibin, alpha S100P 0.461 4p16 S100 calcium binding protein P VPS37A 0.447 8p22 vacuolar protein sorting 37 homolog A (S. cerevisiae) SLC16A14 0.447 2q36.3 solute carrier family 16, member 14 PPARGC1A 0.443 4p15.1 peroxisome proliferator-activated receptor gamma, coactivator 1 alpha SIK1 0.435 21q22.3 salt-inducible kinase 1 IRS2 0.434 13q34 insulin receptor substrate 2 RND1 0.433 12q12 Rho family GTPase 1 HGD 0.433 3q13.33 homogentisate 1,2-dioxygenase PTP4A1 0.432 6q12 protein tyrosine phosphatase type IVA, member 1 C8orf4 0.428 8p11.2 chromosome 8 open reading frame 4 DDC 0.427 7p12.2 dopa decarboxylase (aromatic L-amino acid decarboxylase) TACC2 0.427 10q26 transforming, acidic coiled-coil containing protein 2 MUC13 0.422 3q21.2 mucin 13, cell surface associated C5 0.412 9q33-q34 complement component 5 NR4A2 0.412 2q22-q23 nuclear receptor subfamily 4, group A, member 2 EYS 0.411 6q12 eyes shut homolog (Drosophila) GPX2 0.406 14q24.1 glutathione peroxidase -

Supplementary Materials
Supplementary materials Supplementary Table S1: MGNC compound library Ingredien Molecule Caco- Mol ID MW AlogP OB (%) BBB DL FASA- HL t Name Name 2 shengdi MOL012254 campesterol 400.8 7.63 37.58 1.34 0.98 0.7 0.21 20.2 shengdi MOL000519 coniferin 314.4 3.16 31.11 0.42 -0.2 0.3 0.27 74.6 beta- shengdi MOL000359 414.8 8.08 36.91 1.32 0.99 0.8 0.23 20.2 sitosterol pachymic shengdi MOL000289 528.9 6.54 33.63 0.1 -0.6 0.8 0 9.27 acid Poricoic acid shengdi MOL000291 484.7 5.64 30.52 -0.08 -0.9 0.8 0 8.67 B Chrysanthem shengdi MOL004492 585 8.24 38.72 0.51 -1 0.6 0.3 17.5 axanthin 20- shengdi MOL011455 Hexadecano 418.6 1.91 32.7 -0.24 -0.4 0.7 0.29 104 ylingenol huanglian MOL001454 berberine 336.4 3.45 36.86 1.24 0.57 0.8 0.19 6.57 huanglian MOL013352 Obacunone 454.6 2.68 43.29 0.01 -0.4 0.8 0.31 -13 huanglian MOL002894 berberrubine 322.4 3.2 35.74 1.07 0.17 0.7 0.24 6.46 huanglian MOL002897 epiberberine 336.4 3.45 43.09 1.17 0.4 0.8 0.19 6.1 huanglian MOL002903 (R)-Canadine 339.4 3.4 55.37 1.04 0.57 0.8 0.2 6.41 huanglian MOL002904 Berlambine 351.4 2.49 36.68 0.97 0.17 0.8 0.28 7.33 Corchorosid huanglian MOL002907 404.6 1.34 105 -0.91 -1.3 0.8 0.29 6.68 e A_qt Magnogrand huanglian MOL000622 266.4 1.18 63.71 0.02 -0.2 0.2 0.3 3.17 iolide huanglian MOL000762 Palmidin A 510.5 4.52 35.36 -0.38 -1.5 0.7 0.39 33.2 huanglian MOL000785 palmatine 352.4 3.65 64.6 1.33 0.37 0.7 0.13 2.25 huanglian MOL000098 quercetin 302.3 1.5 46.43 0.05 -0.8 0.3 0.38 14.4 huanglian MOL001458 coptisine 320.3 3.25 30.67 1.21 0.32 0.9 0.26 9.33 huanglian MOL002668 Worenine -

Genomic Analyses of PMBL Reveal New Drivers and Mechanisms of Sensitivity to PD-1 Blockade
Regular Article LYMPHOID NEOPLASIA Genomic analyses of PMBL reveal new drivers and mechanisms of sensitivity to PD-1 blockade Bjoern Chapuy,1,2,* Chip Stewart,3,* Andrew J. Dunford,3,* Jaegil Kim,3 Kirsty Wienand,1 Atanas Kamburov,3 Gabriel K. Griffin,4 Pei-Hsuan Chen,4 Ana Lako,4 Robert A. Redd,5 Claire M. Cote,1 Matthew D. Ducar,6 Aaron R. Thorner,6 Scott J. Rodig,4 Gad Getz,3,7,8,† Downloaded from https://ashpublications.org/blood/article-pdf/134/26/2369/1549702/bloodbld2019002067.pdf by Margaret Shipp on 30 December 2019 and Margaret A. Shipp1,† 1Department of Medical Oncology, Dana-Farber Cancer Institute, Harvard Medical School, Boston, MA; 2Department of Hematology and Oncology, University Medical Center Gottingen,¨ Gottingen,¨ Germany; 3Broad Institute of Harvard and Massachusetts Institute of Technology, Cambridge, MA; 4Department of Pathology, Brigham and Women’s Hospital, Harvard Medical School, Boston, MA; 5Department of Data Sciences and 6Center for Cancer Genome Discovery, Dana-Farber Cancer Institute, Boston, MA; and 7Department of Pathology and 8Center for Cancer Research, Massachusetts General Hospital, Harvard Medical School, Boston, MA KEY POINTS Primary mediastinal large B-cell lymphomas (PMBLs) are aggressive tumors that typically present as large mediastinal masses in young women. PMBLs share clinical, transcriptional, l Comprehensive genomic analyses of and molecular features with classical Hodgkin lymphoma (cHL), including constitutive PMBL reveal new activation of nuclear factor kB (NF-kB), JAK/STAT signaling, and programmed cell death genetic drivers such protein 1 (PD-1)–mediated immune evasion. The demonstrated efficacy of PD-1 blockade in as ZNF217. relapsed/refractory PMBLs led to recent approval by the US Food and Drug Administration l High mutational and underscored the importance of characterizing targetable genetic vulnerabilities in this burden, MSI, and disease. -

Transcriptome Analysis of Human Diabetic Kidney Disease
ORIGINAL ARTICLE Transcriptome Analysis of Human Diabetic Kidney Disease Karolina I. Woroniecka,1 Ae Seo Deok Park,1 Davoud Mohtat,2 David B. Thomas,3 James M. Pullman,4 and Katalin Susztak1,5 OBJECTIVE—Diabetic kidney disease (DKD) is the single cases, mild and then moderate mesangial expansion can be leading cause of kidney failure in the U.S., for which a cure has observed. In general, diabetic kidney disease (DKD) is not yet been found. The aim of our study was to provide an considered a nonimmune-mediated degenerative disease unbiased catalog of gene-expression changes in human diabetic of the glomerulus; however, it has long been noted that kidney biopsy samples. complement and immunoglobulins sometimes can be de- — tected in diseased glomeruli, although their role and sig- RESEARCH DESIGN AND METHODS Affymetrix expression fi arrays were used to identify differentially regulated transcripts in ni cance is not clear (4). 44 microdissected human kidney samples. The DKD samples were The understanding of DKD has been challenged by multi- significant for their racial diversity and decreased glomerular ple issues. First, the diagnosis of DKD usually is made using filtration rate (~20–30 mL/min). Stringent statistical analysis, using clinical criteria, and kidney biopsy often is not performed. the Benjamini-Hochberg corrected two-tailed t test, was used to According to current clinical practice, the development of identify differentially expressed transcripts in control and diseased albuminuria in patients with diabetes is sufficient to make the glomeruli and tubuli. Two different Web-based algorithms were fi diagnosis of DKD (5). We do not understand the correlation used to de ne differentially regulated pathways. -
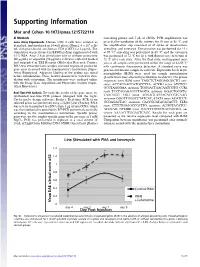
Supporting Information
Supporting Information Mor and Cohen 10.1073/pnas.1215722110 SI Methods containing primer and 5 μL of cDNA. PCR amplification was Gene Array Experiments. Human CD4 T cells were isolated as preceded by incubation of the mixture for 10 min at 95 °C, and described, and incubated in 24-well plates (Nunc), 4 × 106 cells/ the amplification step consisted of 45 cycles of denaturation, mL with plate-bound anti-human CD3 (OKT3) at 2 μg/mL. The annealing, and extension. Denaturation was performed for 15 s stimulation was performed in RPMI medium supplemented with at 95 °C; annealing was performed in 60 °C; and the extension 0.1% BSA. After 2 h of stimulation with or without cefuroxime was performed at 72 °C for 20 s, with fluorescence detection at (50 μg/mL) or ampicillin (50 μg/mL), cells were collected washed 72 °C after each cycle. After the final cycle, melting-point anal- and suspended in TRI Reagent (Molecular Research Center). yses of all samples were performed within the range of 62–95 °C RNA was extracted from samples and used to prepare probes for with continuous fluorescence detection. A standard curve was gene array in accord with the manufacturer’s instructions (Super- generated from one sample in each run. Expression levels of β2- Array Bioscience). Adequate labeling of the probes was tested microglobulin (B2M) were used for sample normalization before hybridization. Three healthy donors were tested in stim- (β-actin levels were affected by cefuroxime treatment). The primer ulation with cefuroxime. The membranes were analyzed online sequences were B2M sense TAGCTCTAGGAGGGCTG anti- with the Image Data Acquisition and Expression Analysis (Super- sense ACCACAACCATGCCTTA; ACVR2 sense ATCTCC- Array Bioscience). -

Genome-Wide Identification of Genes Regulating DNA Methylation Using Genetic Anchors for Causal Inference
bioRxiv preprint doi: https://doi.org/10.1101/823807; this version posted October 30, 2019. The copyright holder for this preprint (which was not certified by peer review) is the author/funder, who has granted bioRxiv a license to display the preprint in perpetuity. It is made available under aCC-BY 4.0 International license. Genome-wide identification of genes regulating DNA methylation using genetic anchors for causal inference Paul J. Hop1,2, René Luijk1, Lucia Daxinger3, Maarten van Iterson1, Koen F. Dekkers1, Rick Jansen4, BIOS Consortium5, Joyce B.J. van Meurs6, Peter A.C. ’t Hoen 7, M. Arfan Ikram8, Marleen M.J. van Greevenbroek9,10, Dorret I. Boomsma11, P. Eline Slagboom1, Jan H. Veldink2, Erik W. van Zwet12, Bastiaan T. Heijmans1* 1 Molecular Epidemiology, Department of Biomedical Data Sciences, Leiden University Medical Center, Leiden, 2333 ZC, The Netherlands 2 Department of Neurology, UMC Utrecht Brain Center, University Medical Centre Utrecht, Utrecht University, Utrecht 3584 CG, Netherlands 3 Department of Human Genetics, Leiden University Medical Center, Leiden, 2333 ZC, The Netherlands 4 Department of Psychiatry, Amsterdam UMC, Vrije Universiteit Amsterdam, Amsterdam Neuroscience, Amsterdam, 1081 HV, The Netherlands 5 Biobank-based Integrated Omics Study Consortium. For a complete list of authors, see the acknowledgements. 6 Department of Internal Medicine, Erasmus Medical Centre, Rotterdam, The Netherlands. 7 Centre for Molecular and Biomolecular Informatics, Radboud Institute for Molecular Life Sciences, Radboud University -

IKBKB Polyclonal Antibody
IKBKB polyclonal antibody Gene Summary: NFKB1 (MIM 164011) or NFKB2 (MIM 164012) is bound to REL (MIM 164910), RELA (MIM Catalog Number: PAB18155 164014), or RELB (MIM 604758) to form the NFKB complex. The NFKB complex is inhibited by I-kappa-B Regulatory Status: For research use only (RUO) proteins (NFKBIA, MIM 164008, or NFKBIB, MIM 604495), which inactivate NF-kappa-B by trapping it in Product Description: Rabbit polyclonal antibody raised the cytoplasm. Phosphorylation of serine residues on the against synthetic peptide of IKBKB. I-kappa-B proteins by kinases (IKBKA, MIM 600664, or IKBKB) marks them for destruction via the ubiquitination Immunogen: A synthetic peptide corresponding to pathway, thereby allowing activation of the NF-kappa-B human IKBKB. complex. Activated NFKB complex translocates into the nucleus and binds DNA at kappa-B-binding motifs such Host: Rabbit as 5-prime GGGRNNYYCC 3-prime or 5-prime Reactivity: Human,Mouse,Rat HGGARNYYCC 3-prime (where H is A, C, or T; R is an A or G purine; and Y is a C or T pyrimidine).[supplied by Applications: ELISA, IHC-P, WB-Ce OMIM] (See our web site product page for detailed applications information) References: 1. NIBP, a novel NIK and IKK(beta)-binding protein that Protocols: See our web site at enhances NF-(kappa)B activation. Hu WH, Pendergast http://www.abnova.com/support/protocols.asp or product JS, Mo XM, Brambilla R, Bracchi-Ricard V, Li F, Walters page for detailed protocols WM, Blits B, He L, Schaal SM, Bethea JR. J Biol Chem. 2005 Aug 12;280(32):29233-41. -

NFKBIB & FBXW11 Protein Protein Interaction Antibody Pair
NFKBIB & FBXW11 Protein Protein Interaction Antibody Pair Catalog # : DI0194 規格 : [ 1 Set ] List All Specification Application Image Product This protein protein interaction antibody pair set comes with two In situ Proximity Ligation Assay (Cell) Description: antibodies to detect the protein-protein interaction, one against the NFKBIB protein, and the other against the FBXW11 protein for use in in situ Proximity Ligation Assay. See Publication Reference below. Reactivity: Human Quality Control Protein protein interaction immunofluorescence result. Testing: Representative image of Proximity Ligation Assay of protein-protein interactions between NFKBIB and FBXW11. HeLa cells were stained with anti-NFKBIB rabbit purified polyclonal antibody 1:1200 and anti- FBXW11 mouse monoclonal antibody 1:50. Each red dot represents the detection of protein-protein interaction complex. The images were analyzed using an optimized freeware (BlobFinder) download from The Centre for Image Analysis at Uppsala University. Supplied Antibody pair set content: Product: 1. NFKBIB rabbit purified polyclonal antibody (20 ug) 2. FBXW11 mouse monoclonal antibody (40 ug) *Reagents are sufficient for at least 30-50 assays using recommended protocols. Storage Store reagents of the antibody pair set at -20°C or lower. Please aliquot Instruction: to avoid repeated freeze thaw cycle. Reagents should be returned to - Page 1 of 4 2016/5/19 20°C storage immediately after use. MSDS: Download Publication Reference 1. An analysis of protein-protein interactions in cross-talk pathways reveals CRKL as a novel prognostic marker in hepatocellular carcinoma. Liu CH, Chen TC, Chau GY, Jan YH, Chen CH, Hsu CN, Lin KT, Juang YL, Lu PJ, Cheng HC, Chen MH, Chang CF, Ting YS, Kao CY, Hsiao M, Huang CY. -

The Role of the NFAT Signalling Pathway in Diffuse Large B-Cell Lymphoma
The role of the NFAT signalling pathway in Diffuse Large B-cell Lymphoma Holly White Doctor of Philosophy Thesis September 2015 Supervisors: Dr Chris Bacon, Professor Neil Perkins & Dr Vikki Rand Northern Institute for Cancer Research Word count: 66,024 1 Abstract Diffuse Large B-Cell Lymphomas (DLBCL) are common, aggressive malignancies of mature B-lymphocytes that represent ~40% of lymphomas. Despite the widespread use of combined immunochemotherapy, approximately 50% of patients with DLBCL die from their disease. The two main DLBCL subgroups resemble activated B cells (ABC) or germinal centre B cells (GCB), where patients with ABC-DLBCL have significantly worse outcome. There is urgent need for novel therapeutic strategies in the treatment of DLBCL, which requires a better understanding of the molecular pathways upon which tumours depend. Accumulating evidence suggests that the signalling networks promoting and sustaining DLBCL derive from dysregulation of the normal pathways controlling B-lymphocyte activation and differentiation. There is increasing evidence indicating important roles for the NFAT family of transcription factors in DLBCL. Constitutively-active nuclear NFAT2 has been demonstrated in approximately 40% of primary DLBCL samples and NFAT has been shown to regulate a small number of genes associated with DLBCL growth/survival. This project investigated the role of NFAT in DLBCL. Nuclear localisation and activation of NFAT family members were characterised in a panel of DLBCL cell lines and chemical inhibition of calcineurin/NFAT, using Cyclosporin A (CsA), indicated dependency on the calcineurin/NFAT pathway for survival. Gene expression microarray analysis performed in DLBCL cell lines treated with CsA revealed potential NFAT target genes involved in the tumour microenvironment and anergy.