NIH Public Access Provided by Cold Spring Harbor Laboratory Institutional Repository Author Manuscript Nat Methods
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Supplementary Data
SUPPLEMENTARY DATA A cyclin D1-dependent transcriptional program predicts clinical outcome in mantle cell lymphoma Santiago Demajo et al. 1 SUPPLEMENTARY DATA INDEX Supplementary Methods p. 3 Supplementary References p. 8 Supplementary Tables (S1 to S5) p. 9 Supplementary Figures (S1 to S15) p. 17 2 SUPPLEMENTARY METHODS Western blot, immunoprecipitation, and qRT-PCR Western blot (WB) analysis was performed as previously described (1), using cyclin D1 (Santa Cruz Biotechnology, sc-753, RRID:AB_2070433) and tubulin (Sigma-Aldrich, T5168, RRID:AB_477579) antibodies. Co-immunoprecipitation assays were performed as described before (2), using cyclin D1 antibody (Santa Cruz Biotechnology, sc-8396, RRID:AB_627344) or control IgG (Santa Cruz Biotechnology, sc-2025, RRID:AB_737182) followed by protein G- magnetic beads (Invitrogen) incubation and elution with Glycine 100mM pH=2.5. Co-IP experiments were performed within five weeks after cell thawing. Cyclin D1 (Santa Cruz Biotechnology, sc-753), E2F4 (Bethyl, A302-134A, RRID:AB_1720353), FOXM1 (Santa Cruz Biotechnology, sc-502, RRID:AB_631523), and CBP (Santa Cruz Biotechnology, sc-7300, RRID:AB_626817) antibodies were used for WB detection. In figure 1A and supplementary figure S2A, the same blot was probed with cyclin D1 and tubulin antibodies by cutting the membrane. In figure 2H, cyclin D1 and CBP blots correspond to the same membrane while E2F4 and FOXM1 blots correspond to an independent membrane. Image acquisition was performed with ImageQuant LAS 4000 mini (GE Healthcare). Image processing and quantification were performed with Multi Gauge software (Fujifilm). For qRT-PCR analysis, cDNA was generated from 1 µg RNA with qScript cDNA Synthesis kit (Quantabio). qRT–PCR reaction was performed using SYBR green (Roche). -

Chromosome 21 Leading Edge Gene Set
Chromosome 21 Leading Edge Gene Set Genes from chr21q22 that are part of the GSEA leading edge set identifying differences between trisomic and euploid samples. Multiple probe set IDs corresponding to a single gene symbol are combined as part of the GSEA analysis. Gene Symbol Probe Set IDs Gene Title 203865_s_at, 207999_s_at, 209979_at, adenosine deaminase, RNA-specific, B1 ADARB1 234539_at, 234799_at (RED1 homolog rat) UDP-Gal:betaGlcNAc beta 1,3- B3GALT5 206947_at galactosyltransferase, polypeptide 5 BACE2 217867_x_at, 222446_s_at beta-site APP-cleaving enzyme 2 1553227_s_at, 214820_at, 219280_at, 225446_at, 231860_at, 231960_at, bromodomain and WD repeat domain BRWD1 244622_at containing 1 C21orf121 240809_at chromosome 21 open reading frame 121 C21orf130 240068_at chromosome 21 open reading frame 130 C21orf22 1560881_a_at chromosome 21 open reading frame 22 C21orf29 1552570_at, 1555048_at_at, 1555049_at chromosome 21 open reading frame 29 C21orf33 202217_at, 210667_s_at chromosome 21 open reading frame 33 C21orf45 219004_s_at, 228597_at, 229671_s_at chromosome 21 open reading frame 45 C21orf51 1554430_at, 1554432_x_at, 228239_at chromosome 21 open reading frame 51 C21orf56 223360_at chromosome 21 open reading frame 56 C21orf59 218123_at, 244369_at chromosome 21 open reading frame 59 C21orf66 1555125_at, 218515_at, 221158_at chromosome 21 open reading frame 66 C21orf7 221211_s_at chromosome 21 open reading frame 7 C21orf77 220826_at chromosome 21 open reading frame 77 C21orf84 239968_at, 240589_at chromosome 21 open reading frame 84 -

Increased Dosage of the Chromosome 21 Ortholog Dyrk1a Promotes Megakaryoblastic Leukemia in a Murine Model of Down Syndrome
Increased dosage of the chromosome 21 ortholog Dyrk1a promotes megakaryoblastic leukemia in a murine model of Down syndrome Sébastien Malinge, … , Sandeep Gurbuxani, John D. Crispino J Clin Invest. 2012;122(3):948-962. https://doi.org/10.1172/JCI60455. Research Article Individuals with Down syndrome (DS; also known as trisomy 21) have a markedly increased risk of leukemia in childhood but a decreased risk of solid tumors in adulthood. Acquired mutations in the transcription factor–encoding GATA1 gene are observed in nearly all individuals with DS who are born with transient myeloproliferative disorder (TMD), a clonal preleukemia, and/or who develop acute megakaryoblastic leukemia (AMKL). Individuals who do not have DS but bear germline GATA1 mutations analogous to those detected in individuals with TMD and DS-AMKL are not predisposed to leukemia. To better understand the functional contribution of trisomy 21 to leukemogenesis, we used mouse and human cell models of DS to reproduce the multistep pathogenesis of DS-AMKL and to identify chromosome 21 genes that promote megakaryoblastic leukemia in children with DS. Our results revealed that trisomy for only 33 orthologs of human chromosome 21 (Hsa21) genes was sufficient to cooperate with GATA1 mutations to initiate megakaryoblastic leukemia in vivo. Furthermore, through a functional screening of the trisomic genes, we demonstrated that DYRK1A, which encodes dual-specificity tyrosine-(Y)-phosphorylation–regulated kinase 1A, was a potent megakaryoblastic tumor–promoting gene that contributed -

Network-Based Method for Drug Target Discovery at the Isoform Level
www.nature.com/scientificreports OPEN Network-based method for drug target discovery at the isoform level Received: 20 November 2018 Jun Ma1,2, Jenny Wang2, Laleh Soltan Ghoraie2, Xin Men3, Linna Liu4 & Penggao Dai 1 Accepted: 6 September 2019 Identifcation of primary targets associated with phenotypes can facilitate exploration of the underlying Published: xx xx xxxx molecular mechanisms of compounds and optimization of the structures of promising drugs. However, the literature reports limited efort to identify the target major isoform of a single known target gene. The majority of genes generate multiple transcripts that are translated into proteins that may carry out distinct and even opposing biological functions through alternative splicing. In addition, isoform expression is dynamic and varies depending on the developmental stage and cell type. To identify target major isoforms, we integrated a breast cancer type-specifc isoform coexpression network with gene perturbation signatures in the MCF7 cell line in the Connectivity Map database using the ‘shortest path’ drug target prioritization method. We used a leukemia cancer network and diferential expression data for drugs in the HL-60 cell line to test the robustness of the detection algorithm for target major isoforms. We further analyzed the properties of target major isoforms for each multi-isoform gene using pharmacogenomic datasets, proteomic data and the principal isoforms defned by the APPRIS and STRING datasets. Then, we tested our predictions for the most promising target major protein isoforms of DNMT1, MGEA5 and P4HB4 based on expression data and topological features in the coexpression network. Interestingly, these isoforms are not annotated as principal isoforms in APPRIS. -

Integrated Functional Genomic Analysis Enables Annotation of Kidney Genome-Wide Association Study Loci
BASIC RESEARCH www.jasn.org Integrated Functional Genomic Analysis Enables Annotation of Kidney Genome-Wide Association Study Loci Karsten B. Sieber,1 Anna Batorsky,2 Kyle Siebenthall,2 Kelly L. Hudkins,3 Jeff D. Vierstra,2 Shawn Sullivan,4 Aakash Sur,4,5 Michelle McNulty,6 Richard Sandstrom,2 Alex Reynolds,2 Daniel Bates,2 Morgan Diegel,2 Douglass Dunn,2 Jemma Nelson,2 Michael Buckley,2 Rajinder Kaul,2 Matthew G. Sampson,6 Jonathan Himmelfarb,7,8 Charles E. Alpers,3,8 Dawn Waterworth,1 and Shreeram Akilesh3,8 Due to the number of contributing authors, the affiliations are listed at the end of this article. ABSTRACT Background Linking genetic risk loci identified by genome-wide association studies (GWAS) to their causal genes remains a major challenge. Disease-associated genetic variants are concentrated in regions con- taining regulatory DNA elements, such as promoters and enhancers. Although researchers have previ- ously published DNA maps of these regulatory regions for kidney tubule cells and glomerular endothelial cells, maps for podocytes and mesangial cells have not been available. Methods We generated regulatory DNA maps (DNase-seq) and paired gene expression profiles (RNA-seq) from primary outgrowth cultures of human glomeruli that were composed mainly of podo- cytes and mesangial cells. We generated similar datasets from renal cortex cultures, to compare with those of the glomerular cultures. Because regulatory DNA elements can act on target genes across large genomic distances, we also generated a chromatin conformation map from freshly isolated human glomeruli. Results We identified thousands of unique regulatory DNA elements, many located close to transcription factor genes, which the glomerular and cortex samples expressed at different levels. -

Risk Stratification of Triple-Negative Breast Cancer with Core Gene
Breast Cancer Research and Treatment (2019) 178:185–197 https://doi.org/10.1007/s10549-019-05366-x EPIDEMIOLOGY Risk stratifcation of triple‑negative breast cancer with core gene signatures associated with chemoresponse and prognosis Eun‑Kyu Kim1 · Ae Kyung Park2 · Eunyoung Ko3 · Woong‑Yang Park4 · Kyung‑Min Lee5 · Dong‑Young Noh5,6 · Wonshik Han5,6 Received: 8 May 2019 / Accepted: 16 July 2019 / Published online: 24 July 2019 © Springer Science+Business Media, LLC, part of Springer Nature 2019 Abstract Purpose Neoadjuvant chemotherapy studies have consistently reported a strong correlation between pathologic response and long-term outcome in triple-negative breast cancer (TNBC). We aimed to defne minimal gene signatures for predicting chemoresponse by a three-step approach and to further develop a risk-stratifcation method of TNBC. Methods The frst step involved the detection of genes associated with resistance to docetaxel in eight TNBC cell lines, leading to identifcation of thousands of candidate genes. Through subsequent second and third step analyses with gene set enrichment analysis and survival analysis using public expression profles, the candidate gene list was reduced to prognostic core gene signatures comprising ten or four genes. Results The prognostic core gene signatures include three up-regulated (CEBPD, MMP20, and WLS) and seven down- regulated genes (ASF1A, ASPSCR1, CHAF1B, DNMT1, GINS2, GOLGA2P5, and SKA1). We further develop a simple risk-stratifcation method based on expression profles of the core genes. Relative expression values of the up-regulated and down-regulated core genes were averaged into two scores, Up and Down scores, respectively; then samples were stratifed by a diagonal line in a xy plot of the Up and Down scores. -

Early Lineage Priming by Trisomy of Erg Leads to Myeloproliferation in a Down Syndrome Model
Early Lineage Priming by Trisomy of Erg Leads to Myeloproliferation in a Down Syndrome Model The Harvard community has made this article openly available. Please share how this access benefits you. Your story matters Citation Ng, A. P., Y. Hu, D. Metcalf, C. D. Hyland, H. Ierino, B. Phipson, D. Wu, et al. 2015. “Early Lineage Priming by Trisomy of Erg Leads to Myeloproliferation in a Down Syndrome Model.” PLoS Genetics 11 (5): e1005211. doi:10.1371/journal.pgen.1005211. http:// dx.doi.org/10.1371/journal.pgen.1005211. Published Version doi:10.1371/journal.pgen.1005211 Citable link http://nrs.harvard.edu/urn-3:HUL.InstRepos:16120953 Terms of Use This article was downloaded from Harvard University’s DASH repository, and is made available under the terms and conditions applicable to Other Posted Material, as set forth at http:// nrs.harvard.edu/urn-3:HUL.InstRepos:dash.current.terms-of- use#LAA RESEARCH ARTICLE Early Lineage Priming by Trisomy of Erg Leads to Myeloproliferation in a Down Syndrome Model Ashley P. Ng1,2*, Yifang Hu1, Donald Metcalf1,2, Craig D. Hyland1, Helen Ierino1, Belinda Phipson1,3,DiWu4,5, Tracey M. Baldwin1, Maria Kauppi1,2, Hiu Kiu1,2, Ladina Di Rago1, Douglas J. Hilton1,2, Gordon K. Smyth1,3, Warren S. Alexander1,2 1 The Walter and Eliza Hall Institute of Medical Research, Parkville, Victoria, Australia, 2 Department of Medical Biology, The University of Melbourne, Parkville, Victoria, Australia, 3 Department of Mathematics and Statistics, The University of Melbourne, Parkville, Victoria, Australia, 4 Centre for Cancer Research, Monash Institute of Medical Research, Monash University, Clayton, Victoria, Australia, 5 Department of Statistics, Harvard University, Cambridge, Massachusetts, United States of America * [email protected] OPEN ACCESS Abstract Citation: Ng AP, Hu Y, Metcalf D, Hyland CD, Ierino Down syndrome (DS), with trisomy of chromosome 21 (HSA21), is the commonest human H, Phipson B, et al. -
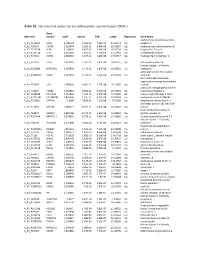
Table S1. the Statistical Metrics for Key Differentially Expressed Genes (Degs)
Table S1. The statistical metrics for key differentially expressed genes (DEGs) Gene Agilent Id Symbol logFC pValue FDR tvalue Regulation Gene Name oxidized low density lipoprotein A_24_P124624 OLR1 2.458429 1.19E-13 7.25E-10 24.04241 Up receptor 1 A_23_P90273 CHST8 2.622464 3.85E-12 6.96E-09 19.05867 Up carbohydrate sulfotransferase 8 A_23_P217528 KLF8 2.109007 4.85E-12 7.64E-09 18.76234 Up Kruppel like factor 8 A_23_P114740 CFH 2.651636 1.85E-11 1.79E-08 17.13652 Up complement factor H A_23_P34031 XAGE2 2.000935 2.04E-11 1.81E-08 17.02457 Up X antigen family member 2 A_23_P27332 TCF4 1.613097 2.32E-11 1.87E-08 16.87275 Up transcription factor 4 histone cluster 1 H1 family A_23_P250385 HIST1H1B 2.298658 2.47E-11 1.87E-08 16.80362 Up member b abnormal spindle microtubule A_33_P3288159 ASPM 2.162032 2.79E-11 2.01E-08 16.66292 Up assembly H19, imprinted maternally expressed transcript (non-protein A_24_P52697 H19 1.499364 4.09E-11 2.76E-08 16.23387 Up coding) potassium voltage-gated channel A_24_P31627 KCNB1 2.289689 6.65E-11 3.97E-08 15.70253 Up subfamily B member 1 A_23_P214168 COL12A1 2.155835 7.59E-11 4.15E-08 15.56005 Up collagen type XII alpha 1 chain A_33_P3271341 LOC388282 2.859496 7.61E-11 4.15E-08 15.55704 Up uncharacterized LOC388282 A_32_P150891 DIAPH3 2.2068 7.83E-11 4.22E-08 15.5268 Up diaphanous related formin 3 zinc finger protein 185 with LIM A_23_P11025 ZNF185 1.385721 8.74E-11 4.59E-08 15.41041 Up domain heat shock protein family B A_23_P96872 HSPB11 1.887166 8.94E-11 4.64E-08 15.38599 Up (small) member 11 A_23_P107454 -

Identification of a Panel of MYC and Tip60 Co-Regulated Genes Functioning Primarily in Cell Cycle and DNA Replication
www.Genes&Cancer.com Genes & Cancer, Vol. 9 (3-4), March 2018 Identification of a panel of MYC and Tip60 co-regulated genes functioning primarily in cell cycle and DNA replication Ling-Jun Zhao1, Paul M. Loewenstein1 and Maurice Green1 1 Department of Microbiology and Molecular Immunology, Saint Louis University School of Medicine, Doisy Research Center, St. Louis, Missouri, USA Correspondence to: Paul M. Loewenstein, email: [email protected] Keywords: MYC; NuA4 complex; Tip60; p300; cancer Received: May 16, 2018 Accepted: July 22, 2018 Published: July 29, 2018 Copyright: Zhao et al. This is an open-access article distributed under the terms of the Creative Commons Attribution License 3.0 (CC BY 3.0), which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited. ABSTRACT We recently reported that adenovirus E1A enhances MYC association with the NuA4/Tip60 histone acetyltransferase (HAT) complex to activate a panel of genes enriched for DNA replication and cell cycle. Genes from this panel are highly expressed in examined cancer cell lines when compared to normal fibroblasts. To further understand gene regulation in cancer by MYC and the NuA4 complex, we performed RNA-seq analysis of MD-MB231 breast cancer cells following knockdown of MYC or Tip60 - the HAT enzyme of the NuA4 complex. We identify here a panel of 424 genes, referred to as MYC-Tip60 co-regulated panel (MTcoR), that are dependent on both MYC and Tip60 for expression and likely co-regulated by MYC and the NuA4 complex. The MTcoR panel is most significantly enriched in genes involved in cell cycle and/ or DNA replication. -

CHAF1A (D77D5) XP Rabbit Mab A
Revision 1 C 0 2 ® - t CHAF1A (D77D5) XP Rabbit mAb a e r o t S Orders: 877-616-CELL (2355) [email protected] Support: 877-678-TECH (8324) 0 8 Web: [email protected] 4 www.cellsignal.com 5 # 3 Trask Lane Danvers Massachusetts 01923 USA For Research Use Only. Not For Use In Diagnostic Procedures. Applications: Reactivity: Sensitivity: MW (kDa): Source/Isotype: UniProt ID: Entrez-Gene Id: WB, IP, IF-IC H Mk Endogenous 145 Rabbit IgG Q13111 10036 Product Usage Information 7. Polo, S.E. et al. (2004) Cancer Res 64, 2371-81. Application Dilution Western Blotting 1:1000 Immunoprecipitation 1:100 Immunofluorescence (Immunocytochemistry) 1:200 Storage Supplied in 10 mM sodium HEPES (pH 7.5), 150 mM NaCl, 100 µg/ml BSA, 50% glycerol and less than 0.02% sodium azide. Store at –20°C. Do not aliquot the antibody. Specificity / Sensitivity CHAF1A (D77D5) XP® Rabbit mAb detects endogenous levels of total CHAF1A protein. Species Reactivity: Human, Monkey Source / Purification Monoclonal antibody is produced by immunizing animals with a synthetic peptide corresponding to residues surrounding Pro303 of human CHAF1A protein. Background Chromatin assembly factor 1 (CAF-1) is a histone H3/H4 chaperone complex that functions in de novo assembly of nucleosomes during DNA replication and nucleotide excision repair (1). Nucleosome assembly is a two-step process, involving initial deposition of a histone H3/H4 tetramer onto DNA, followed by the deposition of a pair of histone H2A/H2B dimers (1). CAF-1 interacts with PCNA and localizes to DNA replication and DNA repair foci, where it functions to assemble newly synthesized histone H3/H4 tetramers onto replicating DNA (2-6). -

Enhanced Autophagic-Lysosomal Activity and Increased BAG3- Mediated Selective Macroautophagy As Adaptive Response of Neuronal Ce
bioRxiv preprint doi: https://doi.org/10.1101/580977; this version posted March 18, 2019. The copyright holder for this preprint (which was not certified by peer review) is the author/funder. All rights reserved. No reuse allowed without permission. Enhanced autophagic-lysosomal activity and increased BAG3- mediated selective macroautophagy as adaptive response of neuronal cells to chronic oxidative stress Debapriya Chakraborty1, Vanessa Felzen1, Christof Hiebel1, Elisabeth Stürner1, Natarajan Perumal2, Caroline Manicam2, Elisabeth Sehn3, Franz Grus2, Uwe Wolfrum3, Christian Behl1* 1Institute of Pathobiochemistry, University Medical Center Mainz of the Johannes Gutenberg University, 55099 Mainz, Germany 2Experimental and Translational Ophthalmology, University Medical Center Mainz, 55131 Mainz, Germany 3 Institute for Molecular Physiology, Johannes Gutenberg University, 55128 Mainz, Germany E-mail addresses of authors: D. Chakraborty: [email protected]; V. Felzen: [email protected]; C. Hiebel: [email protected]; E. Stürner: [email protected]; N. Perumal: nperumal@eye- research.org; C. Manicam: [email protected]; E. Sehn: sehn@uni- mainz.de; F. Grus: [email protected]; U. Wolfrum: [email protected]; *Correspondence to Christian Behl, PhD Institute of Pathobiochemistry University Medical Center of the Johannes Gutenberg University Duesbergweg 6, D-55099 Mainz [email protected] Tel.: +49-6131-39-25890; Fax: +49-6131-39-25792 1 bioRxiv preprint doi: https://doi.org/10.1101/580977; this version posted March 18, 2019. The copyright holder for this preprint (which was not certified by peer review) is the author/funder. All rights reserved. No reuse allowed without permission. Abstract Oxidative stress and a disturbed cellular protein homeostasis (proteostasis) belong to the most important hallmarks of aging and of neurodegenerative disorders. -

Systematic Functional Characterization of Human 21St Chromosome Orthologs In
G3: Genes|Genomes|Genetics Early Online, published on January 24, 2018 as doi:10.1534/g3.118.200019 SECTION: Genome and Systems Biology Systematic Functional Characterization of Human 21st Chromosome Orthologs in Caenorhabditis elegans SARAH K. NORDQUIST*, SOFIA R. SMITH, JONATHAN T. PIERCE *,§ *Institute for Neuroscience, § Institute for Cellular and Molecular Biology, § Center for Learning and Memory, § Waggoner Center for Alcohol & Addiction Research, Department of Neuroscience, The University of Texas at Austin, TX, 78712 1 © The Author(s) 2013. Published by the Genetics Society of America. Short running title: Roles of HSA21 Orthologs in C. elegans Key words: Down syndrome, neuromuscular, neurological, synaptic §To whom correspondence should be addressed: Jonathan T. Pierce University of Texas at Austin Department of Neuroscience 2506 Speedway NMS 5.234 Mailcode C7350 Austin, TX 78712 E-mail: [email protected] Phone: 512-232-4137 2 ABSTRACT Individuals with Down syndrome have neurological and muscle impairments due to an additional copy of the human 21st chromosome (HSA21). Only a few of ~200 HSA21 genes encoding protein have been linked to specific Down syndrome phenotypes, while the remainder are understudied. To identify poorly characterized HSA21 genes required for nervous system function, we studied behavioral phenotypes caused by loss-of- function mutations in conserved HSA21 orthologs in the nematode Caenorhabditis elegans. We identified ten HSA21 orthologs that are required for neuromuscular behaviors: cle-1 (COL18A1), cysl-2 (CBS), dnsn-1 (DONSON), eva-1 (EVA1C), mtq-2 (N6ATM1), ncam-1 (NCAM2), pad-2 (POFUT2), pdxk-1 (PDXK), rnt-1 (RUNX1), and unc-26 (SYNJ1). We also found that three of these genes are required for normal release of the neurotransmitter acetylcholine.