Arhgef12 Drives IL17A-Induced Airway Contractility and Airway Hyperresponsiveness in Mice
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

G Protein Alpha 13 (GNA13) (NM 006572) Human Tagged ORF Clone Lentiviral Particle Product Data
OriGene Technologies, Inc. 9620 Medical Center Drive, Ste 200 Rockville, MD 20850, US Phone: +1-888-267-4436 [email protected] EU: [email protected] CN: [email protected] Product datasheet for RC207762L3V G protein alpha 13 (GNA13) (NM_006572) Human Tagged ORF Clone Lentiviral Particle Product data: Product Type: Lentiviral Particles Product Name: G protein alpha 13 (GNA13) (NM_006572) Human Tagged ORF Clone Lentiviral Particle Symbol: GNA13 Synonyms: G13 Vector: pLenti-C-Myc-DDK-P2A-Puro (PS100092) ACCN: NM_006572 ORF Size: 1131 bp ORF Nucleotide The ORF insert of this clone is exactly the same as(RC207762). Sequence: OTI Disclaimer: The molecular sequence of this clone aligns with the gene accession number as a point of reference only. However, individual transcript sequences of the same gene can differ through naturally occurring variations (e.g. polymorphisms), each with its own valid existence. This clone is substantially in agreement with the reference, but a complete review of all prevailing variants is recommended prior to use. More info OTI Annotation: This clone was engineered to express the complete ORF with an expression tag. Expression varies depending on the nature of the gene. RefSeq: NM_006572.3 RefSeq Size: 4744 bp RefSeq ORF: 1134 bp Locus ID: 10672 UniProt ID: Q14344, A0A024R8M0 Domains: G-alpha Protein Families: Druggable Genome Protein Pathways: Long-term depression, Regulation of actin cytoskeleton, Vascular smooth muscle contraction MW: 44 kDa This product is to be used for laboratory only. Not for diagnostic or therapeutic use. View online » ©2021 OriGene Technologies, Inc., 9620 Medical Center Drive, Ste 200, Rockville, MD 20850, US 1 / 2 G protein alpha 13 (GNA13) (NM_006572) Human Tagged ORF Clone Lentiviral Particle – RC207762L3V Gene Summary: Guanine nucleotide-binding proteins (G proteins) are involved as modulators or transducers in various transmembrane signaling systems (PubMed:15240885, PubMed:16787920, PubMed:16705036, PubMed:27084452). -

PLXNB1 (Plexin
Atlas of Genetics and Cytogenetics in Oncology and Haematology OPEN ACCESS JOURNAL AT INIST-CNRS Gene Section Mini Review PLXNB1 (plexin B1) José Javier Gómez-Román, Montserrat Nicolas Martínez, Servando Lazuén Fernández, José Fernando Val-Bernal Department of Anatomical Pathology, Marques de Valdecilla University Hospital, Medical Faculty, University of Cantabria, Santander, Spain (JJGR, MN, SL, JFVB) Published in Atlas Database: March 2009 Online updated version: http://AtlasGeneticsOncology.org/Genes/PLXNB1ID43413ch3p21.html DOI: 10.4267/2042/44702 This work is licensed under a Creative Commons Attribution-Noncommercial-No Derivative Works 2.0 France Licence. © 2010 Atlas of Genetics and Cytogenetics in Oncology and Haematology Identity Pseudogene No. Other names: KIAA0407; MGC149167; OTTHUMP00000164806; PLEXIN-B1; PLXN5; SEP Protein HGNC (Hugo): PLXNB1 Location: 3p21.31 Description Local order: The Plexin B1 gene is located between 2135 Amino acids (AA). Plexins are receptors for axon FBXW12 and CCDC51 genes. molecular guidance molecules semaphorins. Plexin signalling is important in pathfinding and patterning of both neurons and developing blood vessels. Plexin-B1 is a surface cell receptor. When it binds to its ligand SEMA4D it activates several pathways by binding of cytoplasmic ligands, like RHOA activation and subsequent changes of the actin cytoskeleton, axon guidance, invasive growth and cell migration. It monomers and heterodimers with PLXNB2 after proteolytic processing. Binds RAC1 that has been activated by GTP binding. It binds PLXNA1 and by similarity ARHGEF11, Note ARHGEF12, ERBB2, MET, MST1R, RND1, NRP1 Size: 26,200 bases. and NRP2. Orientation: minus strand. This family features the C-terminal regions of various plexins. The cytoplasmic region, which has been called DNA/RNA a SEX domain in some members of this family is involved in downstream signalling pathways, by Description interaction with proteins such as Rac1, RhoD, Rnd1 and other plexins. -

The Smooth Muscle-Selective Rhogap GRAF3 Is a Critical Regulator of Vascular Tone and Hypertension
ARTICLE Received 9 Jun 2013 | Accepted 11 Nov 2013 | Published 13 Dec 2013 DOI: 10.1038/ncomms3910 The smooth muscle-selective RhoGAP GRAF3 is a critical regulator of vascular tone and hypertension Xue Bai1, Kaitlin C. Lenhart1, Kim E. Bird1, Alisa A. Suen1, Mauricio Rojas2, Masao Kakoki1, Feng Li1, Oliver Smithies1,2, Christopher P. Mack1,2 & Joan M. Taylor1,2 Although hypertension is a worldwide health issue, an incomplete understanding of its aetiology has hindered our ability to treat this complex disease. Here we identify arhgap42 (also known as GRAF3) as a Rho-specific GAP expressed specifically in smooth muscle cells (SMCs) in mice and humans. We show that GRAF3-deficient mice exhibit significant hypertension and increased pressor responses to angiotensin II and endothelin-1; these effects are prevented by treatment with the Rho-kinase inhibitor, Y27632. RhoA activity and myosin light chain phosphorylation are elevated in GRAF3-depleted SMCs in vitro and in vivo, and isolated vessel segments from GRAF3-deficient mice show increased contractility. Taken together, our data indicate that GRAF3-mediated inhibition of RhoA activity in vascular SMCs is necessary for maintaining normal blood pressure homoeostasis. Moreover, these findings provide a potential mechanism for a hypertensive locus recently identified within arhgap42 and provide a foundation for the future development of innovative hypertension therapies. 1 Department of Pathology and Lab Medicine, University of North Carolina, 501 Brinkhous-Bullitt Building CB 7525, Chapel Hill, North Carolina 27599, USA. 2 McAllister Heart Institute, University of North Carolina, Chapel Hill, North Carolina 27599, USA. Correspondence and requests for materials shouldbe addressed to J.M.T. -

A Computational Approach for Defining a Signature of Β-Cell Golgi Stress in Diabetes Mellitus
Page 1 of 781 Diabetes A Computational Approach for Defining a Signature of β-Cell Golgi Stress in Diabetes Mellitus Robert N. Bone1,6,7, Olufunmilola Oyebamiji2, Sayali Talware2, Sharmila Selvaraj2, Preethi Krishnan3,6, Farooq Syed1,6,7, Huanmei Wu2, Carmella Evans-Molina 1,3,4,5,6,7,8* Departments of 1Pediatrics, 3Medicine, 4Anatomy, Cell Biology & Physiology, 5Biochemistry & Molecular Biology, the 6Center for Diabetes & Metabolic Diseases, and the 7Herman B. Wells Center for Pediatric Research, Indiana University School of Medicine, Indianapolis, IN 46202; 2Department of BioHealth Informatics, Indiana University-Purdue University Indianapolis, Indianapolis, IN, 46202; 8Roudebush VA Medical Center, Indianapolis, IN 46202. *Corresponding Author(s): Carmella Evans-Molina, MD, PhD ([email protected]) Indiana University School of Medicine, 635 Barnhill Drive, MS 2031A, Indianapolis, IN 46202, Telephone: (317) 274-4145, Fax (317) 274-4107 Running Title: Golgi Stress Response in Diabetes Word Count: 4358 Number of Figures: 6 Keywords: Golgi apparatus stress, Islets, β cell, Type 1 diabetes, Type 2 diabetes 1 Diabetes Publish Ahead of Print, published online August 20, 2020 Diabetes Page 2 of 781 ABSTRACT The Golgi apparatus (GA) is an important site of insulin processing and granule maturation, but whether GA organelle dysfunction and GA stress are present in the diabetic β-cell has not been tested. We utilized an informatics-based approach to develop a transcriptional signature of β-cell GA stress using existing RNA sequencing and microarray datasets generated using human islets from donors with diabetes and islets where type 1(T1D) and type 2 diabetes (T2D) had been modeled ex vivo. To narrow our results to GA-specific genes, we applied a filter set of 1,030 genes accepted as GA associated. -

Cdc42 and Rac1 Activity Is Reduced in Human Pheochromocytoma and Correlates with FARP1 and ARHGEF1 Expression
234 P Croisé et al. Rho-GTPase activity in human 23:4 281–293 Research pheochromocytoma Cdc42 and Rac1 activity is reduced in human pheochromocytoma and correlates with FARP1 and ARHGEF1 expression Pauline Croisé1, Sébastien Houy1, Mathieu Gand1, Joël Lanoix2, Valérie Calco1, Petra Tóth1, Laurent Brunaud3, Sandra Lomazzi4, Eustache Paramithiotis2, Daniel Chelsky2, Stéphane Ory1,* and Stéphane Gasman1,* Correspondence 1Institut des Neurosciences Cellulaires et Intégratives (INCI), CNRS UPR 3212, Strasbourg, France should be addressed 2Caprion Proteome, Inc., Montréal, Québec, Canada to S Ory or S Gasman 3Service de Chirurgie Digestive, Hépato-bilaire et Endocrinienne, CHRU Nancy, Hôpitaux de Brabois, Email Vandoeuvre les Nancy, France [email protected] or 4Centre de Ressources Biologiques (CRB), CHRU Nancy, Hôpitaux de Brabois, Vandoeuvres les Nancy, France [email protected] *(S Ory and S Gasman contributed equally to this work) Abstract Among small GTPases from the Rho family, Cdc42, Rac, and Rho are well known to mediate Key Words a large variety of cellular processes linked with cancer biology through their ability to cycle f Rho-GTPases between an inactive (GDP-bound) and an active (GTP-bound) state. Guanine nucleotide f pheochromocytoma exchange factors (GEFs) stimulate the exchange of GDP for GTP to generate the activated f mass spectrometry form, whereas the GTPase-activating proteins (GAPs) catalyze GTP hydrolysis, leading to f Rho-GEF Endocrine-Related Cancer Endocrine-Related the inactivated form. Modulation of Rho GTPase activity following altered expression of Rho-GEFs and/or Rho-GAPs has already been reported in various human tumors. However, nothing is known about the Rho GTPase activity or the expression of their regulators in human pheochromocytomas, a neuroendocrine tumor (NET) arising from chromaffin cells of the adrenal medulla. -

ARHGEF12 Regulates Erythropoiesis and Is Involved in Erythroid Regeneration After Ferrata Storti Foundation Chemotherapy in Acute Lymphoblastic Leukemia Patients
Red Cell Biology & its Disorders ARTICLE ARHGEF12 regulates erythropoiesis and is involved in erythroid regeneration after Ferrata Storti Foundation chemotherapy in acute lymphoblastic leukemia patients Yangyang Xie,1* Li Gao,2* Chunhui Xu,3 Liming Chu,3,7 Lei Gao,4 Ruichi Wu,1 Yu Liu,1 Ting Liu,1 Xiao-jian Sun,5 Ruibao Ren,5 Jingyan Tang,1 Yi Zheng,6 Yong Zhou7 and Shuhong Shen1 1Key Lab of Pediatrics Hematology/Oncology, Ministry of Health, Department of Haematologica 2020 Hematology/Oncology, Shanghai Children's Medical Center, Shanghai Jiao Tong University, Volume 105(4):925-936 Shanghai, China; 2Department of Hematology and Oncology, Children's Hospital of Soochow University, Suzhou, China; 3Shanghai Institute of Nutrition and Health, University of Chinese Academy of Sciences, Chinese Academy of Sciences, Shanghai, China; 4CAS Key Laboratory of Genome Sciences and Information, Beijing Institute of Genomics, Beijing, China; 5State Key Laboratory for Medical Genomics, Shanghai Institute of Hematology, Ruijin Hospital, Shanghai, China; 6Division of Experimental Hematology and Cancer Biology, Cincinnati Children's Hospital Research Foundation, Cincinnati, OH, USA and 7CAS Key Laboratory of Tissue Microenvironment and Tumor, Shanghai Institute of Nutrition and Health, University of Chinese Academy of Sciences, Chinese Academy of Sciences, Shanghai, China *YX and LG contributed equally to this work. ABSTRACT ematopoiesis is a finely regulated process in vertebrates under both homeostatic and stress conditions. By whole exome sequencing, we Correspondence: Hstudied the genomics of acute lymphoblastic leukemia (ALL) YI ZHENG patients who needed multiple red blood cell (RBC) transfusions after inten- [email protected] sive chemotherapy treatment. ARHGEF12, encoding a RhoA guanine YONG ZHOU nucleotide exchange factor, was found to be associated with chemothera- [email protected] py-induced anemia by genome-wide association study analyses. -

G-Protein-Coupled Receptor Signaling and Polarized Actin Dynamics Drive
RESEARCH ARTICLE elifesciences.org G-protein-coupled receptor signaling and polarized actin dynamics drive cell-in-cell invasion Vladimir Purvanov, Manuel Holst, Jameel Khan, Christian Baarlink, Robert Grosse* Institute of Pharmacology, University of Marburg, Marburg, Germany Abstract Homotypic or entotic cell-in-cell invasion is an integrin-independent process observed in carcinoma cells exposed during conditions of low adhesion such as in exudates of malignant disease. Although active cell-in-cell invasion depends on RhoA and actin, the precise mechanism as well as the underlying actin structures and assembly factors driving the process are unknown. Furthermore, whether specific cell surface receptors trigger entotic invasion in a signal-dependent fashion has not been investigated. In this study, we identify the G-protein-coupled LPA receptor 2 (LPAR2) as a signal transducer specifically required for the actively invading cell during entosis. We find that 12/13G and PDZ-RhoGEF are required for entotic invasion, which is driven by blebbing and a uropod-like actin structure at the rear of the invading cell. Finally, we provide evidence for an involvement of the RhoA-regulated formin Dia1 for entosis downstream of LPAR2. Thus, we delineate a signaling process that regulates actin dynamics during cell-in-cell invasion. DOI: 10.7554/eLife.02786.001 Introduction Entosis has been described as a specialized form of homotypic cell-in-cell invasion in which one cell actively crawls into another (Overholtzer et al., 2007). Frequently, this occurs between tumor cells such as breast, cervical, or colon carcinoma cells and can be triggered by matrix detachment (Overholtzer et al., 2007), suggesting that loss of integrin-mediated adhesion may promote cell-in-cell invasion. -
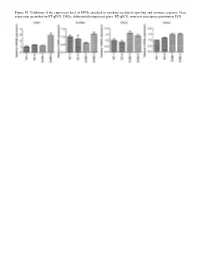
Figure S1. Validation of the Expression Level of Degs Enriched in Cytokine‑Mediated Signaling and Immune Response
Figure S1. Validation of the expression level of DEGs enriched in cytokine‑mediated signaling and immune response. Gene expression quantified by RT‑qPCR. DEGs, differentially expressed genes; RT‑qPCR, reverse‑transcription quantitative PCR. Figure S2. Validation of STAU1‑regulated AS events. IGV‑Sashimi plot revealed (A‑C) three A5SS AS events in three different genes. Reads distribution of each AS event was plotted in the left panel with the transcripts of each gene shown below. The sche‑ matic diagrams depict the structures of two AS events, AS1 (purple line) and AS2 (green line). The exon sequences are denoted by black boxes, the intron sequences by a horizontal line (right panel). RNA‑seq quantification and RT‑qPCR validation of ASEs are presented in the panels on the right. STAU1, double‑stranded RNA‑binding protein Staufen homolog 1; AS, alternative splicing; A5SS, alternative 5'splice site; RNA‑seq, RNA sequencing; RT‑qPCR, reverse‑transcription quantitative PCR. Error bars represent mean ± SEM. *P<0.05. Table SI. Primers used in gene validation experiments. IFIT2‑F CAGCCTACGGCAACTAAA IFIT2‑R GAGCCTTCTCAAAGCACA IFIT3‑F ACACCAAACAATGGCTAC IFIT3‑R TGGACAAACCCTCTAAAC OASL‑F AATGGTGACCGTGATGGG OASL‑R ACCTGAGGATGGAGCAGAG IFI27‑F TTCACTGCGGCGGGAATC IFI27‑R TGGCTGCTATGGAGGACGAG S1PR4‑F TGCTGAAGACGGTGCTGATG S1PR4‑R TGCGGAAGGAGTAGATGATGG CCL5‑F ACGACTGCTGGGTTGGAG CCL5‑R ACCCTGCTGCTTTGCCTA CCL2‑F CTAACCCAGAAACATCCAAT CCL2‑R GCTATGAGCAGCAGGCAC CD44‑F TGGAGGACAGAAAGCCAAGT CD44‑R TTCGCAATGAAACAATCAGTAG PLEKHG2‑M/As‑F CCAAAAGTAAGCCTGTCC PLEKHG2‑M‑R -

High Throughput Strategies Aimed at Closing the GAP in Our Knowledge of Rho Gtpase Signaling
cells Review High Throughput strategies Aimed at Closing the GAP in Our Knowledge of Rho GTPase Signaling Manel Dahmene 1, Laura Quirion 2 and Mélanie Laurin 1,3,* 1 Oncology Division, CHU de Québec–Université Laval Research Center, Québec, QC G1V 4G2, Canada; [email protected] 2 Montréal Clinical Research Institute (IRCM), Montréal, QC H2W 1R7, Canada; [email protected] 3 Université Laval Cancer Research Center, Québec, QC G1R 3S3, Canada * Correspondence: [email protected] Received: 21 May 2020; Accepted: 7 June 2020; Published: 9 June 2020 Abstract: Since their discovery, Rho GTPases have emerged as key regulators of cytoskeletal dynamics. In humans, there are 20 Rho GTPases and more than 150 regulators that belong to the RhoGEF, RhoGAP, and RhoGDI families. Throughout development, Rho GTPases choregraph a plethora of cellular processes essential for cellular migration, cell–cell junctions, and cell polarity assembly. Rho GTPases are also significant mediators of cancer cell invasion. Nevertheless, to date only a few molecules from these intricate signaling networks have been studied in depth, which has prevented appreciation for the full scope of Rho GTPases’ biological functions. Given the large complexity involved, system level studies are required to fully grasp the extent of their biological roles and regulation. Recently, several groups have tackled this challenge by using proteomic approaches to map the full repertoire of Rho GTPases and Rho regulators protein interactions. These studies have provided in-depth understanding of Rho regulators specificity and have contributed to expand Rho GTPases’ effector portfolio. Additionally, new roles for understudied family members were unraveled using high throughput screening strategies using cell culture models and mouse embryos. -

Download (PDF)
Table S1. Putative miR-322 target transcripts which is highly expressed in MGCs ________________________________________________________________________________ 0610037L13Rik, 1700037H04Rik, 2310061I04Rik, 2810006K23Rik, Abcc5, Abhd16a, Acbd3, Acox1, Acsbg1, Acsl4, Actr1a, Actr2, Adck5, Adh5, Adrbk1, Aff4, Agk, Ahcyl1, Akap11, Akap7, Akirin1, Alg3, Amfr, Ammecr1, Amotl2, Ankfy1, Ankhd1, Ankrd52, Ap2a1, Ap2b1, Ap3b1, Ap3d1, App, Arcn1, Arf3, Arfgap2, Arhgap12, Arhgap5, Arhgdia, Arhgef11, Arih1, Arl2, Arl3, Arl8b, Armcx6, Asap1, Asnsd1, Atf6, Atg13, Atg4b, Atp13a3, Atp5g1, Atp6v1a, Atxn2, Atxn7l3, Atxn7l3b, AW549877, B4galt1, B4galt7, Bace1, Bag5, Baiap2, Baz2a, BC037034, Bcl2l1, Bcl2l2, Bfar, Bmpr1a, Bptf, Brd2, Brd4, Brpf3, Btbd3, Btg2, Cab39, Cacna2d1, Calm1, Capns1, Caprin1, Capza2, Carm1, Caskin1, Cbfa2t3, Cbx5, Cbx6, Cc2d1b, Ccdc127, Ccdc6, Ccnd2, Ccnt2, Ccnyl1, Cd164, Cd2ap, Cdc25a, Cdc27, Cdc37l1, Cdc42se2, Cdca4, Cdipt, Cdk5rap3, Cdk8, Cdk9, Cdv3, Celf1, Cfl2, Chchd3, Chd6, Chmp1a, Chmp7, Chordc1, Chpf, Chpt1, Chst8, Chtf8, Cisd2, Clasrp, Clcn3, Clstn1, Cmpk1, Cnih2, Cnot1, Cnot2, Col4a3bp, Cope, Cops2, Cops7a, Cops7b, Copz1, Coq6, Cpd, Crebzf, Crim1, Crk, Crkl, Csde1, Cse1l, Ctnnb1, Cul4a, Cul4b, Cxx1a, Cxx1b, Cxx1c, D15Ertd621e, D2hgdh, Dcaf7, Dcbld2, Dcp1a, Dctn5, Ddost, Ddr1, Ddx39, Ddx3x, Ddx6, Dedd, Dhdds, Dhx16, Diap1, Dido1, Dlst, Dmtf1, Dnaja2, Dnajb14, Dnajb2, Dnajc1, Dnajc16, Dnajc25, Dph3, Dpm1, Dpp9, Dpy19l4, Dsel, Dtl, Dvl1, Dync1li2, Dynll2, Dynlt3, Dyrk1a, Dyrk1b, Ebna1bp2, Edc4, Eftud2, Egln2, Eif1a, Eif2b2, Eif2s1, -

4 Understanding the Role of GNA13 Deregulation in Lymphomagenesis
Integrative Genomics Reveals a Role for GNA13 in Lymphomagenesis by Adrienne Greenough University Program in Genetics and Genomics Duke University Approved: ___________________________ Sandeep Dave, Supervisor ___________________________ Fred Dietrich ___________________________ Jack Keene ___________________________ Yuan Zhuang Dissertation submitted in partial fulfillment of the requirements for the degree of Doctor of Philosophy in the University Program in Genetics and Genomics in the Graduate School of Duke University 2014 i v ABSTRACT Integrative Genomics Reveals a Role for GNA13 in Lymphomagenesis by Adrienne Greenough University Program in Genetics and Genomics Duke University Approved: ___________________________ Sandeep Dave, Supervisor ___________________________ Fred Dietrich ___________________________ Jack Keene ___________________________ Yuan Zhuang An abstract of a dissertation submitted in partial fulfillment of the requirements for the degree of Doctor of Philosophy in the University Program in Genetics and Genomics in the Graduate School of Duke University 2014 Copyright by Adrienne Greenough 2014 Abstract Lymphomas comprise a diverse group of malignancies derived from immune cells. High throughput sequencing has recently emerged as a powerful and versatile method for analysis of the cancer genome and transcriptome. As these data continue to emerge, the crucial work lies in sorting through the wealth of information to hone in on the critical aspects that will give us a better understanding of biology and new insight for how to treat disease. Finding the important signals within these large data sets is one of the major challenges of next generation sequencing. In this dissertation, I have developed several complementary strategies to describe the genetic underpinnings of lymphomas. I begin with developing a better method for RNA sequencing that enables strand-specific total RNA sequencing and alternative splicing profiling in the same analysis. -

Gene Networks Activated by Specific Patterns of Action Potentials in Dorsal Root Ganglia Neurons Received: 10 August 2016 Philip R
www.nature.com/scientificreports OPEN Gene networks activated by specific patterns of action potentials in dorsal root ganglia neurons Received: 10 August 2016 Philip R. Lee1,*, Jonathan E. Cohen1,*, Dumitru A. Iacobas2,3, Sanda Iacobas2 & Accepted: 23 January 2017 R. Douglas Fields1 Published: 03 March 2017 Gene regulatory networks underlie the long-term changes in cell specification, growth of synaptic connections, and adaptation that occur throughout neonatal and postnatal life. Here we show that the transcriptional response in neurons is exquisitely sensitive to the temporal nature of action potential firing patterns. Neurons were electrically stimulated with the same number of action potentials, but with different inter-burst intervals. We found that these subtle alterations in the timing of action potential firing differentially regulates hundreds of genes, across many functional categories, through the activation or repression of distinct transcriptional networks. Our results demonstrate that the transcriptional response in neurons to environmental stimuli, coded in the pattern of action potential firing, can be very sensitive to the temporal nature of action potential delivery rather than the intensity of stimulation or the total number of action potentials delivered. These data identify temporal kinetics of action potential firing as critical components regulating intracellular signalling pathways and gene expression in neurons to extracellular cues during early development and throughout life. Adaptation in the nervous system in response to external stimuli requires synthesis of new gene products in order to elicit long lasting changes in processes such as development, response to injury, learning, and memory1. Information in the environment is coded in the pattern of action-potential firing, therefore gene transcription must be regulated by the pattern of neuronal firing.