Pressure-Stabilized Lithium Caesides with Caesium Anions Beyond the À 1 State
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Atomic Spectra of Alkali Elements
Atomic Spectra of Alkali Elements S. R. Kulkarni April 10, 2020 Rydberg primarily focused on studying the lines of alkali metals (Lithium, Potassium and Sodium).1 Rydberg organized the various features by their appearance on the pho- tographs:The alkali spectra were more complicated than that of hydrogen. Rydberg recog- nized that were three different types of lines: lines which looked \sharp" (on photographic plates), \principal" (strong lines that showed up in emission and absorption) and those which appeared ‘diffuse”. These series were abbreviated to S, P, D. Later \Fundamental" (F) was added. As noted earlier, Rydberg preferred to work with wavenumbers. Using data from Liveing and Deware he recast Angstrom's formula as follows: N k = k − (1) n 1 (n + µ)2 where kn is the wavenumber of the nth line in a given series. Rydberg kept N fixed to the value measured by Balmer with k1 µ being free parameters. Rydberg found the following formulae for Lithium R ks = ks − ; 2; 3; 4; ::: (2) n 1 (n + S)2 R kp = kp − 1; 2; 3; ::: (3) n 1 (n + P )2 R kd = kd − 2; 3; 4; ::: (4) n 1 (n + D)2 s −1 p −1 where S = 0:5951, P = 0:9596, D = 0:9974, k1 = 28601:6 cm k1 = 43487:7 cm , d −1 k1 = 28598:5 cm and we have switched to the modern notation in which N is replaced by R (the value he found was R = 109721:6 cm−1). Rydberg had confidence in the data that he was able to find a deeper connection between the constants between the series. -

The Development of the Periodic Table and Its Consequences Citation: J
Firenze University Press www.fupress.com/substantia The Development of the Periodic Table and its Consequences Citation: J. Emsley (2019) The Devel- opment of the Periodic Table and its Consequences. Substantia 3(2) Suppl. 5: 15-27. doi: 10.13128/Substantia-297 John Emsley Copyright: © 2019 J. Emsley. This is Alameda Lodge, 23a Alameda Road, Ampthill, MK45 2LA, UK an open access, peer-reviewed article E-mail: [email protected] published by Firenze University Press (http://www.fupress.com/substantia) and distributed under the terms of the Abstract. Chemistry is fortunate among the sciences in having an icon that is instant- Creative Commons Attribution License, ly recognisable around the world: the periodic table. The United Nations has deemed which permits unrestricted use, distri- 2019 to be the International Year of the Periodic Table, in commemoration of the 150th bution, and reproduction in any medi- anniversary of the first paper in which it appeared. That had been written by a Russian um, provided the original author and chemist, Dmitri Mendeleev, and was published in May 1869. Since then, there have source are credited. been many versions of the table, but one format has come to be the most widely used Data Availability Statement: All rel- and is to be seen everywhere. The route to this preferred form of the table makes an evant data are within the paper and its interesting story. Supporting Information files. Keywords. Periodic table, Mendeleev, Newlands, Deming, Seaborg. Competing Interests: The Author(s) declare(s) no conflict of interest. INTRODUCTION There are hundreds of periodic tables but the one that is widely repro- duced has the approval of the International Union of Pure and Applied Chemistry (IUPAC) and is shown in Fig.1. -

High-Temperature Structural Evolution of Caesium and Rubidium Triiodoplumbates D.M
High-temperature structural evolution of caesium and rubidium triiodoplumbates D.M. Trots, S.V. Myagkota To cite this version: D.M. Trots, S.V. Myagkota. High-temperature structural evolution of caesium and rubidium tri- iodoplumbates. Journal of Physics and Chemistry of Solids, Elsevier, 2009, 69 (10), pp.2520. 10.1016/j.jpcs.2008.05.007. hal-00565442 HAL Id: hal-00565442 https://hal.archives-ouvertes.fr/hal-00565442 Submitted on 13 Feb 2011 HAL is a multi-disciplinary open access L’archive ouverte pluridisciplinaire HAL, est archive for the deposit and dissemination of sci- destinée au dépôt et à la diffusion de documents entific research documents, whether they are pub- scientifiques de niveau recherche, publiés ou non, lished or not. The documents may come from émanant des établissements d’enseignement et de teaching and research institutions in France or recherche français ou étrangers, des laboratoires abroad, or from public or private research centers. publics ou privés. Author’s Accepted Manuscript High-temperature structural evolution of caesium and rubidium triiodoplumbates D.M. Trots, S.V. Myagkota PII: S0022-3697(08)00173-X DOI: doi:10.1016/j.jpcs.2008.05.007 Reference: PCS 5491 To appear in: Journal of Physics and www.elsevier.com/locate/jpcs Chemistry of Solids Received date: 31 January 2008 Revised date: 2 April 2008 Accepted date: 14 May 2008 Cite this article as: D.M. Trots and S.V. Myagkota, High-temperature structural evolution of caesium and rubidium triiodoplumbates, Journal of Physics and Chemistry of Solids, doi:10.1016/j.jpcs.2008.05.007 This is a PDF file of an unedited manuscript that has been accepted for publication. -

Helium Adsorption on Lithium Substrates
JLowTempPhys DOI 10.1007/s10909-007-9516-5 Helium Adsorption on Lithium Substrates E. Van Cleve · P. Taborek · J.E. Rutledge Received: 25 July 2007 / Accepted: 13 September 2007 © Springer Science+Business Media 2007 Abstract We have developed a cryogenic pulsed laser deposition (PLD) system to deposit lithium films onto a quartz crystal microbalance (QCM) at 4 K. Adsorption isotherms of 4He on lithium were measured in the temperature range between 1.42 K and 2.5 K. The isotherms are qualitatively different from isotherms on strong sub- strates such as gold and weak substrates such as cesium. There is no evidence of the formation of solid-like layers of helium, and the helium coverage is approximately linear in the pressure over a wide range. By measuring the low coverage slope of the isotherms, the binding energy of helium to lithium was found to be approxi- mately −13.6 K. For lithium substrates less than approximately 100 layers thick, the chemical potential at which the superfluid transition was observed was surprisingly sensitive to the details of lithium deposition. Keywords Helium films · Pulsed laser deposition · Superfluidity · Alkali metal 1 Introduction When helium is adsorbed onto a strong heterogenous substrate such as gold, the first 2 or 3 statistical layers are solid-like. The nature of these layers is not yet clear, but the layers are amorphous and do not participate significantly in superflow at high coverages. Superfluidity on strong substrates requires a minimum critical coverage to saturate the solid-like layers, and the superfluid phase which forms at higher cover- ages flows over these layers and does not interact directly with the strong, short range This work was supported by NSF grant DMR 0509685. -

Improvement in Retention of Solid Fission Products in HTGR Fuel Particles by Ceramic Kernel Additives
FORMAL REPORT GERHTR-159 UNITED STATES-GERMAN HIGH TEMPERATURE REACTOR RESEARCH EXCHANGE PROGRAM Original report number ______________________ Title Improvement in Retention of Solid Fission Products in HTGR Fuel Particles by Ceramic Kernel Additives Authorial R. Forthmann, E. Groos and H. Grobmeier Originating Installation Kemforschtmgsanlage Juelich, West Germany. Date of original report issuance August 1975_______ Reporting period covered _ _____________________________ In the original English This report, translated wholly or in part from the original language, has been reproduced directly from copy pre pared by the United States Mission to the European Atomic Energy Community THIS REPORT MAY BE GIVEN UNLIMITED DISTRIBUTION ERDA Technical Information Center, Oak Ridge, Tennessee DISCLAIMER Portions of this document may be illegible in electronic image products. Images are produced from the best available original document. GERHTR-159 Distribution Category UC-77 CONTENTS page 1. INTRODUCTION 2 2. FUNDAMENTAL STUDIES 3 3. IRRADIATION EXPERIMENT FRJ2-P17 5 3.1 Results of the Fission Product 8 Release Measurements Improvement in Retention of Solid 3.2 Electron Microprobe Investigations Fission Products in HTGR Fuel Particles 8 by Ceramic Kernel Additives. 4. IRRADIATION EXPERIMENT FRJ2-P18 16 4.1 Release of Solid Fission Products 19 4.2 Electron Microprobe Studies 24 by R. Forthmann, E. Groos, H. GrObmeier 5. SUMMARY AND CONCLUSIONS 27 6. ACKNOWLEDGEMENT 28 7. REFERENCES 29 2 - X. INTRODUCTION Kernforschungs- anlage JUTich JOL - 1226 August 1975 Considerations of the core design of advanced High-Temperature Gas-cooled GmbH IRW Reactors (HTGRs) led to increased demands concerning solid fission product retention in the fuel elements. This would be desirable not only for HTGR power plants with a helium-turbine in the primary circuit (HHT project), but also for the application of HTGRs as a source of nuclear process heat. -

Heavier Alkali Metal Complexes of 2-Phenylamidopyridine: an X- Ray Crystallographic and Theoretical Study of a Structurally Diverse Series of Crown Ether Adducts
Edinburgh Research Explorer Heavier alkali metal complexes of 2-phenylamidopyridine: An X- ray crystallographic and theoretical study of a structurally diverse series of crown ether adducts Citation for published version: Morrison, C, Liddle, ST & Clegg, W 2004, 'Heavier alkali metal complexes of 2-phenylamidopyridine: An X- ray crystallographic and theoretical study of a structurally diverse series of crown ether adducts', Dalton Transactions, no. 16, pp. 2514-2525. https://doi.org/10.1039/B406741J Digital Object Identifier (DOI): 10.1039/B406741J Link: Link to publication record in Edinburgh Research Explorer Document Version: Peer reviewed version Published In: Dalton Transactions Publisher Rights Statement: Copyright © 2004 by the Royal Society of Chemistry. All rights reserved. General rights Copyright for the publications made accessible via the Edinburgh Research Explorer is retained by the author(s) and / or other copyright owners and it is a condition of accessing these publications that users recognise and abide by the legal requirements associated with these rights. Take down policy The University of Edinburgh has made every reasonable effort to ensure that Edinburgh Research Explorer content complies with UK legislation. If you believe that the public display of this file breaches copyright please contact [email protected] providing details, and we will remove access to the work immediately and investigate your claim. Download date: 04. Oct. 2021 Post-print of a peer-reviewed article published by the Royal Society of Chemistry. Published article available at: http://dx.doi.org/10.1039/B406741J Cite as: Morrison, C., Liddle, S. T., & Clegg, W. (2004). Heavier alkali metal complexes of 2- phenylamidopyridine: An X-ray crystallographic and theoretical study of a structurally diverse series of crown ether adducts. -

Lithium Data Sheet
98 LITHIUM (Data in metric tons of lithium content unless otherwise noted) Domestic Production and Use: The only lithium production in the United States was from a brine operation in Nevada. Two companies produced a wide range of downstream lithium compounds in the United States from domestic or imported lithium carbonate, lithium chloride, and lithium hydroxide. Domestic production data were withheld to avoid disclosing company proprietary data. Although lithium markets vary by location, global end-use markets are estimated as follows: batteries, 65%; ceramics and glass, 18%; lubricating greases, 5%; polymer production, 3%; continuous casting mold flux powders, 3%; air treatment, 1%; and other uses, 5%. Lithium consumption for batteries has increased significantly in recent years because rechargeable lithium batteries are used extensively in the growing market for portable electronic devices and increasingly are used in electric tools, electric vehicles, and grid storage applications. Lithium minerals were used directly as ore concentrates in ceramics and glass applications. Salient Statistics—United States: 2015 2016 2017 2018 2019e Production W W W W W Imports for consumption 2,750 3,140 3,330 3,420 2,500 Exports 1,790 1,520 1,960 1,660 1,700 Consumption, estimated1 2,000 3,000 3,000 3,000 2,000 Price, annual average, battery-grade lithium carbonate, dollars per metric ton2 6,500 8,650 15,000 17,000 13,000 Employment, mine and mill, number 70 70 70 70 70 Net import reliance3 as a percentage of estimated consumption >25 >50 >50 >50 >25 Recycling: One domestic company has recycled lithium metal and lithium-ion batteries since 1992 at its facility in British Columbia, Canada. -

Density Functional Theory Metadynamics of Silver, Caesium and Palladium Diffusion at B-Sic Grain Boundaries ⇑ Jeremy Rabone A, , Eddie López-Honorato B
Journal of Nuclear Materials 458 (2015) 56–63 Contents lists available at ScienceDirect Journal of Nuclear Materials journal homepage: www.elsevier.com/locate/jnucmat Density functional theory metadynamics of silver, caesium and palladium diffusion at b-SiC grain boundaries ⇑ Jeremy Rabone a, , Eddie López-Honorato b a European Commission, Joint Research Centre, Institute for Transuranium Elements, D-76125 Karlsruhe, Germany b Centro de Investigación y de Estudios Avanzados del IPN (CINVESTAV), Unidad Saltillo, Industria Metalúrgica 1062, Parque Industrial, Ramos Arizpe 25900, Coahuila, Mexico highlights DFT metadynamics of diffusion of Pd, Ag and Cs on grain boundaries in b-SiC. The calculated diffusion rates for Pd and Ag tally with experimental release rates. A mechanism of release other than grain boundary diffusion seems likely for Cs. article info abstract Article history: The use of silicon carbide in coated nuclear fuel particles relies on this materials impermeability towards Received 22 May 2014 fission products under normal operating conditions. Determining the underlying factors that control the Accepted 9 November 2014 rate at which radionuclides such as Silver-110m and Caesium-137 can cross the silicon carbide barrier Available online 4 December 2014 layers, and at which fission products such as palladium could compromise or otherwise alter the nature of this layer, are of paramount importance for the safety of this fuel. To this end, DFT-based metadynam- ics simulations are applied to the atomic diffusion of silver, caesium and palladium along a R5 grain boundary and to palladium along a carbon-rich R3 grain boundary in cubic silicon carbide at 1500 K. For silver, the calculated diffusion coefficients lie in a similar range (7.04 Â 10À19–3.69 Â 10À17 m2 sÀ1) as determined experimentally. -

Biologists Struggle with Push to End Use of Caesium Proposed Switch to X-Ray Irradiators Could Affect Results of Research
NEWS IN FOCUS NUCLEAR SECURITY Biologists struggle with push to end use of caesium Proposed switch to X-ray irradiators could affect results of research. BY JEFF TOLLEFSON nybody who wants to conduct experiments on mice in Margaret Goodell’s immunology lab must sub- Amit to a host of security measures, starting JIM R. BOUNDS/AP/PA with a background check by the FBI. That’s because Goodell, a researcher at Baylor College of Medicine in Houston, Texas, uses a caesium-based irradiator to destroy bone marrow in mice that are set to receive stem- cell transplants. The US government fears that the radioactive caesium could be stolen to make a ‘dirty’ bomb. Now the US National Nuclear Security Administration (NNSA) is working with sci- entists to investigate how — or whether — to replace caesium irradiators with less dangerous X-ray technology. Researchers have used the caesium devices for decades, to study every thing from immunotherapy to cancer treat- ment, and some fear that switching to X-ray irradiators will affect their results. Goodell, who has found subtle differences in how the mouse immune system responds Biomedical researchers often use caesium-137 to irradiate cells. to the two types of device, prefers Baylor’s caesium irradiator. Her research has revealed or water; exposure to the substance can cause alternative ways to treat blood is relatively that immune cells called B lymphocytes burns, radiation sickness or death, depend- simple. The NNSA is working with research- recovered more slowly in mice treated with ing on the dose. Caesium irradiators, which ers to pin down the more complicated issue an X-ray irradiator than in those exposed have long been used to eliminate pathogens in of how X-ray irradiators might differ from to caesium. -

Expansion of Domestic Production of Lithium Carbonate and Lithium Hydroxide to Supply US Battery Industry
Expansion of Domestic Production of Lithium Carbonate and Lithium Hydroxide to Supply US Battery Industry John Groves Jeff Davis Chemetall Foote Corp May 11, 2011 Project ID# ARRAVT010 This presentation does not contain any proprietary, confidential, or otherwise restricted information Overview Expand Lithium Raw Material Base in US Timeline Barriers Start Date: April 14, 2010 Geothermal Resource Strength and Viability of End Date: February, 2013 Geothermal resource Partners Budget Engineering: BE&K® (a KBR DOE Share - $28.4 million company) Rockwood Share - $39.5 Environmental Assessment: million Nevada Bureau of Land Mgmt Relevance: Domestic Source of Strategic Materials • Objectives – Expand domestic lithium carbonate and lithium hydroxide production to supply the US electric drive automotive market. – Deliver high quality lithium products to battery component manufacturers to produce high quality lithium ion batteries. – Create construction jobs over the first two years in the US and permanent jobs for production of lithium raw materials. – Stimulate the US economy with worthwhile long term benefits that will support the conversion to electric drive mobility. Relevance: Domestic Source of Strategic Materials • Milestones – Deliver battery grade lithium products to the DOE and component manufacturers in 2012 from this project. – Maintain the long term viability of domestic production of lithium raw materials by lowering operating cost and at the same time reducing fossil fuel based energy consumption. – Job Creation throughout 2010-2012 -

8. Chemistry of the Main Group Elements Unusual Bonding
8. Chemistry of the Main Group Elements A Snapshot on Main Group Chemistry unusual bonding , structure & reactivity 8. Chemistry of the Main Group Elements A Snapshot on Main Group Chemistry very powerful reducing agent te 2+ a − in d r o ? o ! c n - o b six r a c 2− Na2 [Ne]3s2 {Ba-cryptand} + disodide 2− M.Y. Redko et al. JACS 2003 gold(I) methanium also, in NH3(l) H. Schmidbaur et al. Na + (NH ) e − Chem. Ber. 1992 3 x 1 8. Chemistry of the Main Group Elements A Snapshot on Main Group Chemistry very powerful reducing agent 2+ − 2− Na2 [Ne]3s2 {Ba-cryptand} + disodide 2− M.Y. Redko et al. JACS 2003 gold(I) methanium also, in NH3(l) H. Schmidbaur & F. Gabbai Na + (NH ) e − Chem. Ber. 1997 3 x 8. Chemistry of the Main Group Elements here we go again… table salt #1 …well, not in my book! Check out Nitrogenase or Cytochrome C-Oxidase…or Hemoglobin… 2 8. Chemistry of the Main Group Elements Hemoglobin 8. Chemistry of the Main Group Elements more on that later… 3 8. Chemistry of the Main Group Elements General Trends in Main Group Chemistry Electrical Resistivities: far right: non-metals pnic(t)ogens (pnigo = choke), chalcogens, halogens & noble gases middle: C: Diamond, graphite & fullerenes Si: Silicon, Ge: germanium, Sn & Pb far left: metals alkali metals & alkaline earths: luster, high ability to conduct heat & electricity, malleability 8. Chemistry of the Main Group Elements General Trends in Main Group Chemistry Electrical Resistivities: Carbon conductivity 154.5 pm parallel to layers: σ = C-C 154 pm 2 C=C 134 pm 3 2.6 x 104 sp sp Ω-1cm-1 + pπz T ¼, σ ¿ metal conductivity perp. -
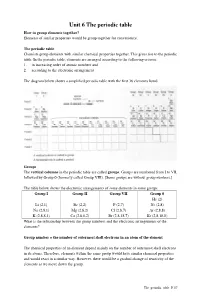
Unit 6 the Periodic Table How to Group Elements Together? Elements of Similar Properties Would Be Group Together for Convenience
Unit 6 The periodic table How to group elements together? Elements of similar properties would be group together for convenience. The periodic table Chemists group elements with similar chemical properties together. This gives rise to the periodic table. In the periodic table, elements are arranged according to the following criteria: 1. in increasing order of atomic numbers and 2. according to the electronic arrangement The diagram below shows a simplified periodic table with the first 36 elements listed. Groups The vertical columns in the periodic table are called groups . Groups are numbered from I to VII, followed by Group 0 (formerly called Group VIII). [Some groups are without group numbers.] The table below shows the electronic arrangements of some elements in some groups. Group I Group II Group VII Group 0 He (2) Li (2,1) Be (2,2) F (2,7) Ne (2,8) Na (2,8,1) Mg (2,8,2) Cl (2,8,7) Ar (2,8,8) K (2,8,8,1) Ca (2,8,8,2) Br (2,8,18,7) Kr (2,8,18,8) What is the relationship between the group numbers and the electronic arrangements of the elements? Group number = the number of outermost shell electrons in an atom of the element The chemical properties of an element depend mainly on the number of outermost shell electrons in its atoms. Therefore, elements within the same group would have similar chemical properties and would react in a similar way. However, there would be a gradual change of reactivity of the elements as we move down the group.