The Genomic Makeup of Nine Horse Populations Sampled in the Netherlands
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Population Genetic Analysis of the Estonian Native Horse Suggests Diverse and Distinct Genetics, Ancient Origin and Contribution from Unique Patrilines
G C A T T A C G G C A T genes Article Population Genetic Analysis of the Estonian Native Horse Suggests Diverse and Distinct Genetics, Ancient Origin and Contribution from Unique Patrilines Caitlin Castaneda 1 , Rytis Juras 1, Anas Khanshour 2, Ingrid Randlaht 3, Barbara Wallner 4, Doris Rigler 4, Gabriella Lindgren 5,6 , Terje Raudsepp 1,* and E. Gus Cothran 1,* 1 College of Veterinary Medicine and Biomedical Sciences, Texas A&M University, College Station, TX 77843, USA 2 Sarah M. and Charles E. Seay Center for Musculoskeletal Research, Texas Scottish Rite Hospital for Children, Dallas, TX 75219, USA 3 Estonian Native Horse Conservation Society, 93814 Kuressaare, Saaremaa, Estonia 4 Institute of Animal Breeding and Genetics, University of Veterinary Medicine Vienna, 1210 Vienna, Austria 5 Department of Animal Breeding and Genetics, Swedish University of Agricultural Sciences, 75007 Uppsala, Sweden 6 Livestock Genetics, Department of Biosystems, KU Leuven, B-3001 Leuven, Belgium * Correspondence: [email protected] (T.R.); [email protected] (E.G.C.) Received: 9 August 2019; Accepted: 13 August 2019; Published: 20 August 2019 Abstract: The Estonian Native Horse (ENH) is a medium-size pony found mainly in the western islands of Estonia and is well-adapted to the harsh northern climate and poor pastures. The ancestry of the ENH is debated, including alleged claims about direct descendance from the extinct Tarpan. Here we conducted a detailed analysis of the genetic makeup and relationships of the ENH based on the genotypes of 15 autosomal short tandem repeats (STRs), 18 Y chromosomal single nucleotide polymorphisms (SNPs), mitochondrial D-loop sequence and lateral gait allele in DMRT3. -

List of Horse Breeds 1 List of Horse Breeds
List of horse breeds 1 List of horse breeds This page is a list of horse and pony breeds, and also includes terms used to describe types of horse that are not breeds but are commonly mistaken for breeds. While there is no scientifically accepted definition of the term "breed,"[1] a breed is defined generally as having distinct true-breeding characteristics over a number of generations; its members may be called "purebred". In most cases, bloodlines of horse breeds are recorded with a breed registry. However, in horses, the concept is somewhat flexible, as open stud books are created for developing horse breeds that are not yet fully true-breeding. Registries also are considered the authority as to whether a given breed is listed as Light or saddle horse breeds a "horse" or a "pony". There are also a number of "color breed", sport horse, and gaited horse registries for horses with various phenotypes or other traits, which admit any animal fitting a given set of physical characteristics, even if there is little or no evidence of the trait being a true-breeding characteristic. Other recording entities or specialty organizations may recognize horses from multiple breeds, thus, for the purposes of this article, such animals are classified as a "type" rather than a "breed". The breeds and types listed here are those that already have a Wikipedia article. For a more extensive list, see the List of all horse breeds in DAD-IS. Heavy or draft horse breeds For additional information, see horse breed, horse breeding and the individual articles listed below. -

ASHA of NEW YORK Presents the 47Th SYRACUSE INTERNATIONAL HORSE SHOW
ASHA OF NEW YORK Presents The 47th SYRACUSE INTERNATIONAL HORSE SHOW To Benefit Upstate New York Hospice Alliance June 24-27, 2020 Closing Date: May 22, 2020 New York State Fairgrounds www.ashany.org 1 Notes 2 SYRACUSE INTERNATIONAL HORSE SHOW June 24-27, 2020 New York State Fairgrounds, Syracuse, NY OFFICIALS The following have been invited to officiate. Show Management reserves the right to change or substitute officials. Judges.................................................................................................Lewis Eckard, Hildebran, NC Saddlebred, Saddle Seat Equitation, Hackney, Roadster, Miscellaneous …………………………………………………………………… Tom Bombolis, Stillwater, MN Morgan, Morgan Equitation, Dutch Harness Horse, Opportunity, Miscellaneous Academy Judge……………………………………………………………….………………TBD Steward……………………………………………………………....…Stacey Wright, Blairs, VA Manager....................................................................................................Liz Holmes, Mebane, NC (919-672-3741, [email protected] Show Secretary............................................... ................................Linda Burke, Horseheads, NY (607) 739-6169 [email protected] Announcer.............................................................................................Wyatt Lilly, Princeton, WV Ringmasters............................................................................................Don Baker, Louisville, KY John Franzreb, Staten Island, NY Veterinarian............................................................Finger -
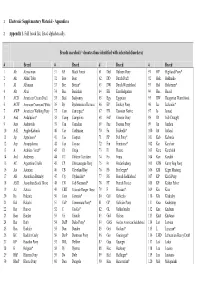
Electronic Supplementary Material - Appendices
1 Electronic Supplementary Material - Appendices 2 Appendix 1. Full breed list, listed alphabetically. Breeds searched (* denotes those identified with inherited disorders) # Breed # Breed # Breed # Breed 1 Ab Abyssinian 31 BF Black Forest 61 Dul Dülmen Pony 91 HP Highland Pony* 2 Ak Akhal Teke 32 Boe Boer 62 DD Dutch Draft 92 Hok Hokkaido 3 Al Albanian 33 Bre Breton* 63 DW Dutch Warmblood 93 Hol Holsteiner* 4 Alt Altai 34 Buc Buckskin 64 EB East Bulgarian 94 Huc Hucul 5 ACD American Cream Draft 35 Bud Budyonny 65 Egy Egyptian 95 HW Hungarian Warmblood 6 ACW American Creme and White 36 By Byelorussian Harness 66 EP Eriskay Pony 96 Ice Icelandic* 7 AWP American Walking Pony 37 Cam Camargue* 67 EN Estonian Native 97 Io Iomud 8 And Andalusian* 38 Camp Campolina 68 ExP Exmoor Pony 98 ID Irish Draught 9 Anv Andravida 39 Can Canadian 69 Fae Faeroes Pony 99 Jin Jinzhou 10 A-K Anglo-Kabarda 40 Car Carthusian 70 Fa Falabella* 100 Jut Jutland 11 Ap Appaloosa* 41 Cas Caspian 71 FP Fell Pony* 101 Kab Kabarda 12 Arp Araappaloosa 42 Cay Cayuse 72 Fin Finnhorse* 102 Kar Karabair 13 A Arabian / Arab* 43 Ch Cheju 73 Fl Fleuve 103 Kara Karabakh 14 Ard Ardennes 44 CC Chilean Corralero 74 Fo Fouta 104 Kaz Kazakh 15 AC Argentine Criollo 45 CP Chincoteague Pony 75 Fr Frederiksborg 105 KPB Kerry Bog Pony 16 Ast Asturian 46 CB Cleveland Bay 76 Fb Freiberger* 106 KM Kiger Mustang 17 AB Australian Brumby 47 Cly Clydesdale* 77 FS French Saddlebred 107 KP Kirdi Pony 18 ASH Australian Stock Horse 48 CN Cob Normand* 78 FT French Trotter 108 KF Kisber Felver 19 Az Azteca -

2016 Brittish Riding Pony Breed Standards and Showing Rules
2016 Brittish Riding Pony Breed Standards and Showing Rules Secretary: Mrs Alicia M Hay Blairview, By Milnathort, KY13 0SF 07970 816416 www.npsscotland.co.uk BRITISH RIDING PONY BREED DESCRIPTION The British Riding Pony is a breed, established over a hundred years ago, originally by the Polo Pony Stud Book Society in 1893. However, 20 years later it became the National Pony Society and to this day it is the custodian of the Stud Book. The Stud Book was formed to encourage the breeding, registration and improvement of both Riding Ponies and at that time all the native breds too, though they now have their own stud books. The foundation blood lines of all the British Riding Ponies were Polo Ponies, Thoroughbreds, Arabs and the British Native Breeds (mostly Welsh or Dartmoor). An increasing number of the ponies now being registered with the NPS are the progeny of British Riding Pony sires and dams and through many generations of selective breeding a very high standard has been achieved. British Riding Ponies are of three categories or types – Show Ponies, Show Hunter Ponies and Sports/Competition Ponies. The Sports Ponies result from cross breeding with Sport Horses or Ponies. All types have outstanding quality while retaining the pony characteristics of good temperament, hardiness, soundness and surefootedness. They provide an ideal mount for today’s competitive riders and are successful in a wide variety of equine competitions and disciplines. The British Riding Pony is much respected and sought after world wide and some of the best blood lines have been exported, predominantly to Australia, New Zealand and America. -
The Commercial & Savings Bank the CSB MOBILE APP the App That Makes a Snap
THE CSB MOBILE APP Your local CSB is now in the palm The App of your hand! Pay bills, check account balances, make deposits and transfer That Makes funds on-the-go with our new 1 Banking CSB mobile banking app. The CSB app is available for download A Snap. at csb1.com or in app stores now. The Commercial & Savings Bank 330-674-9015 1-800-654-9015 www.csb .com | | 1 Serving you with Banking Centers in Millersburg, Berlin, Charm, Gnadenhutten, New Philadelphia, North Canton, Orrville, Shreve, Sugarcreek, Walnut Creek, Winesburg and Wooster. 2 Consigned to Carriage Sale #31 Road Grader 3 TENTATIVE AUCTION SCHEDULE tuesdaY: 8:00 A.M. Tack Sale 9:00 A.M. Horse Drawn Equipment & Field Related Items sold on outside All field items and equipment must be checked in by monday evening 10:00 a.m. - Antiques followed by saddles - New Arena Harnesses Sell Tuesday, 10:00 a.m. in Main Ring followed by Collars on outside 12:00 Noon Box Wagons on outside 1:00 P.M. - All Carriages will sell through the ring. 100 carriages consigned prior to publishing of catalog. (Note: Unloading and checking in for Tuesday’s sale can be done Friday, Saturday, and Monday, Feb. 28, March 1 and 3). 4:00 P.M. - NEW - Pony Pull for consigned ponies 5:00 P.M. - Trail Horse Demonstration for Cataloged Horses on the Obstacle Course 5:00 P.M. - Ponies wednesdaY 8:00 A.M. - Crossbreds, Spotted Drafts and Riding Horses. thursdaY 7:30 A.M. - Horse Pull Demonstration for Cataloged and Uncataloged Horses. -

01622 633060 Kent Showground, Maidstone ME14
www.kentshow.co.uk 01622 633060 Kent Showground, Maidstone ME14 3JF KENT COUNTY AGRICULTURAL SOCIETY Patron: HRH The Duke of Kent, KG President: The Lord Colgrain, DL Chairman: Mr K Attwood OFFICIAL SCHEDULE OF THE EIGHTY NINTH KENT COUNTY SHOW FRIDAY - SATURDAY - SUNDAY 06 07 08 JULY 2018 ENTRIES CLOSE Cattle, Sheep, Wool and Goats - Friday 18 May 2018 Horses and Ponies - Friday 18 May 2018 Show Jumping - Friday 22 June 2018 SHOWGROUND HOLDING NUMBER 20/059/8000 Please send your entries to the Livestock and Equine Department Kent County Agricultural Society Kent Showground Detling, Maidstone Kent ME14 3JF Tel: 01622 630975 www.kentshowground.co.uk This Schedule is issued subject to the Rules, Orders and Regulations of the Department for the Environment, Food & Rural Affairs 1 PRESIDENTS AND DEPUTY PRESIDENTS PAST AND PRESENT 1923 The Right Honourable THE EARL OF DARNLEY, CA, JP, DL 1924-1925 R BRUCE WARD 1926 Capt J I H FRIEND, OBE, MC, JP, DL 1927 The Right Honourable LORD SACKVILLE, CBE, JP, DL 1928-1929 The Right Honourable THE EARL OF RADNOR, KG, KCVO 1930 The Right Honourable SIR PHILIP SASSOON, Bart, PC, MP 1931 Major G WHELER, MC 1932 Col The Right Honourable LORD CORNWALLIS CBE, JP, DL 1933 The Right Honourable LORD PLENDER, CBE, LLD, JP 1934-1935 R BRUCE WARD 1936-1937 W K WHIGHAM, JP 1938 The Right Honourable THE EARL OF RADNOR, KG, KCVO 1939 C TUFF, DL, JP, (Sir Charles Tuff) 1947-1981 The Right Honourable LORD CORNWALLIS, KCVO, KBE, MC 1947-1965 SIR EDWARD HARDY (Deputy President) 1966-1971 SIR LESLIE DOUBLEDAY JP (Deputy -

Association of the DMRT3 Nonsense Mutation with Pattern of Locomotion in Five Different Horse Breeds
Swedish University of Agricultural Sciences Faculty of Veterinary Medicine and Animal Science Association of the DMRT3 nonsense mutation with pattern of locomotion in five different horse breeds Niina Kangas Examensarbete / Swedish University of Agricultural Sciences Master Thesis, 30 hp Department of Animal Breeding and Genetics Agriculture Programme 408 – Animal Science Uppsala 2013 Swedish University of Agricultural Sciences Faculty of Veterinary Medicine and Animal Science Department of Animal Breeding and Genetics Association of the DMRT3 nonsense mutation with pattern of locomotion in five different horse breeds Association av stoppmutationen i DMRT3 med rörelsemönster hos fem olika hästraser Niina Kangas Supervisors: Lisa Andersson, SLU, Department of Animal Breeding and Genetics Gabriella Lindgren, SLU, Department of Animal Breeding and Genetics Examiner: Susanne Eriksson, SLU, Department of Animal Breeding and Genetics Credits: 30 hp Course title: Degree project in Animal Science Course code: EX0558 Programme: Agriculture Programme – Animal Science Level: Advanced, A2E Place of publication: Uppsala Year of publication: 2013 Cover picture: Upper left (Coldblooded trotter), lower right (Standardbred): Kim Jäderkvist Upper right (American Curly): Jak Curly, France Middle (Icelandic Horse): Lennart Lindholm Lower left (Morgan): Sara Huhtanen Persons and horses on cover picture did not have anything to do with the study They only illustrate the horse breeds in this study Name of series: Examensarbete / Swedish University of Agricultural -

The Ambling Influence.Pdf
THE AMBLING INFLUENCE end up in the ASB PART 1 The American Saddlebred Horse is famous for his Cave drawings from the Steppes of Asia (http://www.spanishjennet.org/history.shtml). gaits, but where do these gaits come from? Gaited horses have been around for many years, but how did they end up in the American Saddlebred? This series of articles will take you from the dawn of the gaited horse through to the modern day Saddlebred, look at the genetics behind the ambling gait and give you some pointers as to the physique of the gaited horse. What is a gaited horse anyway? Every pace of the horse, be it walk, trot or canter, is called a “gait”. For the gaited enthusiast, any horse can do these gaits, what they are interested in is the smooth non-jarring English palfrey, cc 1795 – 1865. lateral gait (the legs on one side moving together). (http://www.1st-art-gallery.com/John- This “gait” comes in many guises and names Frederick-Herring-Snr/My-Ladye's-Palfrey.html). depending on the collection, speed and length of stride of the horse, as well as the individual breed of the horse. It is the specific pattern of footfall and the cadence that defines the gait in each of the gaited breeds. A quiet horse may well have a better gait than his flashy fast-moving counterpart, so look beyond the hype and see exactly what those feet and hindquarters are doing. This smooth-moving gait has been depicted in cave walls and fossilised in footprints dating to over 3½ million years ago – so just how did it get from there Lady Conaway's Spanish Jennet to the American Saddlebred? We know that horses (http://www.spanishjennet.org/registry.shtml) are not native to America, so to answer that question we must travel back in time and place to Europe and Asia. -

For Immediate Release for More Information, Contact Ginny Simon, Project Marketing, 610-889-2036, [email protected]
For Immediate Release for more information, contact Ginny Simon, Project Marketing, 610-889-2036, [email protected] September 24, 2019 (Devon, PA) - It doesn't get much cuter than watching foals as they first experience the Dixon Oval. But it's especially fun when it's under the shining sun! Individual Breed Classes From the adorable Haflingers to the impressive Hanoverians and Oldenbergs, more than 15 breeds showed attendees what careful breeding can do to develop and strengthen a breed. They were all winners but the top three scorers were: Kandyvrowe (Glocks Toto Jr. x Fergievrouwe), a 2015 mare bred by M.Hendrikden-de-Rijk, and owned by Iron Spring Hanoverian Farm, earned the high score of 81.850 Gypsy ISF (Tjalbert 460 x Goaitske fan e, a Friesian bred by Iron Spring Farm, scored a 80.60 Sophistikate, a Hanovarien handled by Francine Gentile, was third high scorer with a score of 79.250. Many of the breeds - - Hanoverians, Oldenburgs, Dutch Warmbloods are familiar favorites. Others, while well-loved an appreciated by their owners and breeders, are not so well known. Take the Dutch Harness Horse for example. Also known as “Tuigpaard," Dutch Harness Horses are a warmblood breed of fine driving horses that began in the Netherlands after World War II. These work horses primarily worked in farms and fields; however, horse owners became intensely competitive and desired to breed the finest and most elegant horse as horses were a status symbol to farmers when they would visit the town. As a result, breeders aimed to produce a refined, high-stepping horse that are easily recognizable. -

The Kerry Bog Pony Brochure
The Kerry Bog Pony www.kerrybogpony.ie Photo: Bob Langrish THE KERRY BOG PONY The Kerry Bog Pony is a small mountain and moorland type pony. The breed originated in Kerry but it is now found all over Ireland. The numbers however are very low and the breed is still critically ▲ Photo: Liz Sugar endangered. It has a fine intelligent head with large kind eyes. It has a strong and well set on neck with a rounded shoulder and compact body. The pony is clean legged with very little feather to its heels. It has good bone, with short cannon bones, short pasterns and upright hooves. The Kerry Bog Pony is extremely hardy, resistant to many equine diseases with great powers of endurance. Its temperament and versatility make it an excellent children’s pony and it can be used ▲ Photo: Gay Keogh by adults for carriage driving and as a pack animal. Though an ancient breed it was only officially recognised by the Department of Agriculture and the European Union in 2004. Photo: Pascal Lando BREED STANDARD SIZE: This is a small pony evolved as such because of its use as a draught animal in the bogs of Kerry over the centuries. Thus, the height of the Kerry Bog pony is 102 cms - 117 cms for Stallions and Geldings and 102 cms - 112cms for Mares. COLOUR: Any strong whole colour is to be found, but colour is generally brown or brownish black and bay. Some chestnut, grey and dun colours are also to be found. COAT: The coat of the Kerry Bog Pony is long and dense and easily capable of withstanding harsh winter conditions without shelter. -

Wild Horse DNA Report
! ! LEGAL COVENANT FROM THE XENI GWET'IN GOVERNMENT !!!!!!!!!!!!!! in the lands described in , 2013 SC C 44, and their Aboriginal rights to hunt and trap throughout the area claimed in Nation v. British Columbia rights to hunt and trap birds and animals for the purposes of securing animals for work and transportation, food, clothing, shelter, mats, blankets, and crafts, as well as for spiritual, ceremonial, and cultural uses throughout the Brittany T riangle ( This right is inclusive of a right to capture and use horses for transportation and work. The Claim A rea is within the m A rea. Nothing said in our meetings or documents shall abrogate or derogate from Tsilh Del, esqox. ! 2! Characteristic*wild*horse*pocket/wetland*sedge/grassland*habitat*of*the*Brittany*Triangle*Plateau.*This*is*one*of*the*most*remote*and* harsh*wild*horse*areas*left*in*Canada.*This*is*an*unusually*large*group*of*wild*horses,*as*bands*in*the*Brittany*Triangle*usually*number*10@ 14*horses.*Chris*Harris*photo.* * ! 3! Thanks are expressed for financial support from The Vancouver Foundation, Friends of Nemaiah Valley (FONV), Valhalla Wilderness Society (VWS), anonymous donors, and others. Thanks are also extended to the genetics lab at the Department of Veterinary Integrative Bioscience, Texas A&M University, for doing the genetic analysis at nominal costs. research in their Caretaker and Rights Area. Special thanks to Chief Roger William and former Chief Marilyn Baptiste for their ongoing advice and support. BC Parks is thanked for providing research permits for our main sample area, Nunsti Provincial Park. David Williams and Pat Swift of FONV are particularly thanked for their tireless support, enduring faith in the Nemiah People and their horse culture, and for holding so many things together that make things work, as well as for their generosity in providing a comfortable and always interesting research station at Far Meadow.