Thesis Fads2 Expression Modulates Effect Of
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Stearoyl-Coa Desaturase 1 As a Therapeutic Target for the Treatment of Cancer
cancers Review Stearoyl-CoA Desaturase 1 as a Therapeutic Target for the Treatment of Cancer Zuzanna Tracz-Gaszewska and Pawel Dobrzyn * Laboratory of Molecular Medical Biochemistry, Nencki Institute of Experimental Biology Polish Academy of Sciences, 02-093 Warsaw, Poland * Correspondence: [email protected] Received: 5 June 2019; Accepted: 3 July 2019; Published: 5 July 2019 Abstract: A distinctive feature of cancer cells of various origins involves alterations of the composition of lipids, with significant enrichment in monounsaturated fatty acids. These molecules, in addition to being structural components of newly formed cell membranes of intensely proliferating cancer cells, support tumorigenic signaling. An increase in the expression of stearoyl-CoA desaturase 1 (SCD1), the enzyme that converts saturated fatty acids to D9-monounsaturated fatty acids, has been observed in a wide range of cancer cells, and this increase is correlated with cancer aggressiveness and poor outcomes for patients. Studies have demonstrated the involvement of SCD1 in the promotion of cancer cell proliferation, migration, metastasis, and tumor growth. Many studies have reported a role for this lipogenic factor in maintaining the characteristics of cancer stem cells (i.e., the population of cells that contributes to cancer progression and resistance to chemotherapy). Importantly, both the products of SCD1 activity and its direct impact on tumorigenic pathways have been demonstrated. Based on these findings, SCD1 appears to be a significant player in the development of malignant disease and may be a promising target for anticancer therapy. Numerous chemical compounds that exert inhibitory effects on SCD1 have been developed and preclinically tested. The present review summarizes our current knowledge of the ways in which SCD1 contributes to the progression of cancer and discusses opportunities and challenges of using SCD1 inhibitors for the treatment of cancer. -

Supplementary Table S4. FGA Co-Expressed Gene List in LUAD
Supplementary Table S4. FGA co-expressed gene list in LUAD tumors Symbol R Locus Description FGG 0.919 4q28 fibrinogen gamma chain FGL1 0.635 8p22 fibrinogen-like 1 SLC7A2 0.536 8p22 solute carrier family 7 (cationic amino acid transporter, y+ system), member 2 DUSP4 0.521 8p12-p11 dual specificity phosphatase 4 HAL 0.51 12q22-q24.1histidine ammonia-lyase PDE4D 0.499 5q12 phosphodiesterase 4D, cAMP-specific FURIN 0.497 15q26.1 furin (paired basic amino acid cleaving enzyme) CPS1 0.49 2q35 carbamoyl-phosphate synthase 1, mitochondrial TESC 0.478 12q24.22 tescalcin INHA 0.465 2q35 inhibin, alpha S100P 0.461 4p16 S100 calcium binding protein P VPS37A 0.447 8p22 vacuolar protein sorting 37 homolog A (S. cerevisiae) SLC16A14 0.447 2q36.3 solute carrier family 16, member 14 PPARGC1A 0.443 4p15.1 peroxisome proliferator-activated receptor gamma, coactivator 1 alpha SIK1 0.435 21q22.3 salt-inducible kinase 1 IRS2 0.434 13q34 insulin receptor substrate 2 RND1 0.433 12q12 Rho family GTPase 1 HGD 0.433 3q13.33 homogentisate 1,2-dioxygenase PTP4A1 0.432 6q12 protein tyrosine phosphatase type IVA, member 1 C8orf4 0.428 8p11.2 chromosome 8 open reading frame 4 DDC 0.427 7p12.2 dopa decarboxylase (aromatic L-amino acid decarboxylase) TACC2 0.427 10q26 transforming, acidic coiled-coil containing protein 2 MUC13 0.422 3q21.2 mucin 13, cell surface associated C5 0.412 9q33-q34 complement component 5 NR4A2 0.412 2q22-q23 nuclear receptor subfamily 4, group A, member 2 EYS 0.411 6q12 eyes shut homolog (Drosophila) GPX2 0.406 14q24.1 glutathione peroxidase -

Chemical Agent and Antibodies B-Raf Inhibitor RAF265
Supplemental Materials and Methods: Chemical agent and antibodies B-Raf inhibitor RAF265 [5-(2-(5-(trifluromethyl)-1H-imidazol-2-yl)pyridin-4-yloxy)-N-(4-trifluoromethyl)phenyl-1-methyl-1H-benzp{D, }imidazol-2- amine] was kindly provided by Novartis Pharma AG and dissolved in solvent ethanol:propylene glycol:2.5% tween-80 (percentage 6:23:71) for oral delivery to mice by gavage. Antibodies to phospho-ERK1/2 Thr202/Tyr204(4370), phosphoMEK1/2(2338 and 9121)), phospho-cyclin D1(3300), cyclin D1 (2978), PLK1 (4513) BIM (2933), BAX (2772), BCL2 (2876) were from Cell Signaling Technology. Additional antibodies for phospho-ERK1,2 detection for western blot were from Promega (V803A), and Santa Cruz (E-Y, SC7383). Total ERK antibody for western blot analysis was K-23 from Santa Cruz (SC-94). Ki67 antibody (ab833) was from ABCAM, Mcl1 antibody (559027) was from BD Biosciences, Factor VIII antibody was from Dako (A082), CD31 antibody was from Dianova, (DIA310), and Cot antibody was from Santa Cruz Biotechnology (sc-373677). For the cyclin D1 second antibody staining was with an Alexa Fluor 568 donkey anti-rabbit IgG (Invitrogen, A10042) (1:200 dilution). The pMEK1 fluorescence was developed using the Alexa Fluor 488 chicken anti-rabbit IgG second antibody (1:200 dilution).TUNEL staining kits were from Promega (G2350). Mouse Implant Studies: Biopsy tissues were delivered to research laboratory in ice-cold Dulbecco's Modified Eagle Medium (DMEM) buffer solution. As the tissue mass available from each biopsy was limited, we first passaged the biopsy tissue in Balb/c nu/Foxn1 athymic nude mice (6-8 weeks of age and weighing 22-25g, purchased from Harlan Sprague Dawley, USA) to increase the volume of tumor for further implantation. -

Suppression of FADS1 Induces ROS Generation, Cell Cycle Arrest, and Apoptosis in Melanocytes: Implications for Vitiligo
www.aging-us.com AGING 2019, Vol. 11, No. 24 Research Paper Suppression of FADS1 induces ROS generation, cell cycle arrest, and apoptosis in melanocytes: implications for vitiligo Luyan Tang1, Jian Li1, Wenwen Fu1, Wenyu Wu1, Jinhua Xu1 1Department of Dermatology, Huashan Hospital Affiliated to Fudan University, Shanghai 200040, China Correspondence to: Jinhua Xu; email: [email protected] Keywords: vitiligo, microarray, fatty acid desaturase 1, apoptosis, melanocyte Received: May 6, 2019 Accepted: November 7, 2019 Published: December 21, 2019 Copyright: Tang et al. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY 3.0), which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited. ABSTRACT Vitiligo is a potentially serious condition characterized by loss of melanin and death of melanocytes. To identify potential therapeutic targets for vitiligo, we conducted a microarray analysis of three human vitiligo specimens and paired adjacent normal tissues. Because we found that the fatty acid desaturase 1 (FADS1) gene was downregulated in vitiligo specimens, we carried out experiments to assess its role in melanocyte replication and survival. RT-qPCR was used to verify that FADS1 expression was lower in vitiligo-affected tissues and vitiligo melanocyte PIG3V cells than in matched controls or normal human epidermal PIG1 melanocytes. In addition, CCK-8, immunofluorescence, western blot and flow cytometry assay were used to detect the proliferation and apoptosis in PIG1 cells respectively. Overexpression of FADS1 promoted proliferation of PIG3V melanocytes, while FADS1 silencing inhibited proliferation and induced cell death in PIG1 melanocytes. -

Genetic Analyses of Human Fetal Retinal Pigment Epithelium Gene Expression Suggest Ocular Disease Mechanisms
ARTICLE https://doi.org/10.1038/s42003-019-0430-6 OPEN Genetic analyses of human fetal retinal pigment epithelium gene expression suggest ocular disease mechanisms Boxiang Liu 1,6, Melissa A. Calton2,6, Nathan S. Abell2, Gillie Benchorin2, Michael J. Gloudemans 3, 1234567890():,; Ming Chen2, Jane Hu4, Xin Li 5, Brunilda Balliu5, Dean Bok4, Stephen B. Montgomery 2,5 & Douglas Vollrath2 The retinal pigment epithelium (RPE) serves vital roles in ocular development and retinal homeostasis but has limited representation in large-scale functional genomics datasets. Understanding how common human genetic variants affect RPE gene expression could elu- cidate the sources of phenotypic variability in selected monogenic ocular diseases and pin- point causal genes at genome-wide association study (GWAS) loci. We interrogated the genetics of gene expression of cultured human fetal RPE (fRPE) cells under two metabolic conditions and discovered hundreds of shared or condition-specific expression or splice quantitative trait loci (e/sQTLs). Co-localizations of fRPE e/sQTLs with age-related macular degeneration (AMD) and myopia GWAS data suggest new candidate genes, and mechan- isms by which a common RDH5 allele contributes to both increased AMD risk and decreased myopia risk. Our study highlights the unique transcriptomic characteristics of fRPE and provides a resource to connect e/sQTLs in a critical ocular cell type to monogenic and complex eye disorders. 1 Department of Biology, Stanford University, Stanford, CA 94305, USA. 2 Department of Genetics, Stanford University School of Medicine, Stanford, CA 94305, USA. 3 Program in Biomedical Informatics, Stanford University School of Medicine, Stanford 94305 CA, USA. 4 Department of Ophthalmology, Jules Stein Eye Institute, UCLA, Los Angeles 90095 CA, USA. -

Links Between Iron and Lipids: Implications in Some Major Human Diseases
pharmaceuticals Review Links Between Iron and Lipids: Implications in Some Major Human Diseases Stephanie Rockfield, Ravneet Chhabra, Michelle Robertson, Nabila Rehman, Richa Bisht and Meera Nanjundan * Department of Cell Biology, Microbiology and Molecular Biology, University of South Florida, Tampa, FL 336200, USA; srockfi[email protected] (S.R.); [email protected] (R.C.); [email protected] (M.R.); [email protected] (N.R.); [email protected] (R.B.) * Correspondence: [email protected]; Tel.: +1-813-974-8133 Received: 31 August 2018; Accepted: 19 October 2018; Published: 22 October 2018 Abstract: Maintenance of iron homeostasis is critical to cellular health as both its excess and insufficiency are detrimental. Likewise, lipids, which are essential components of cellular membranes and signaling mediators, must also be tightly regulated to hinder disease progression. Recent research, using a myriad of model organisms, as well as data from clinical studies, has revealed links between these two metabolic pathways, but the mechanisms behind these interactions and the role these have in the progression of human diseases remains unclear. In this review, we summarize literature describing cross-talk between iron and lipid pathways, including alterations in cholesterol, sphingolipid, and lipid droplet metabolism in response to changes in iron levels. We discuss human diseases correlating with both iron and lipid alterations, including neurodegenerative disorders, and the available evidence regarding the potential mechanisms underlying how iron may promote disease pathogenesis. Finally, we review research regarding iron reduction techniques and their therapeutic potential in treating patients with these debilitating conditions. We propose that iron-mediated alterations in lipid metabolic pathways are involved in the progression of these diseases, but further research is direly needed to elucidate the mechanisms involved. -

The Key Roles of Elongases and Desaturases in Mammalian Fatty Acid Metabolism: Insights from Transgenic Mice
Progress in Lipid Research 49 (2010) 186–199 Contents lists available at ScienceDirect Progress in Lipid Research journal homepage: www.elsevier.com/locate/plipres Review The key roles of elongases and desaturases in mammalian fatty acid metabolism: Insights from transgenic mice Hervé Guillou a, Damir Zadravec b, Pascal G.P. Martin a, Anders Jacobsson b,* a Integrative Toxicology and Metabolism, Pôle de Toxicologie Alimentaire, Laboratoire de Pharmacologie et Toxicologie, Institut National de la Recherche Agronomique INRA UR66, Toulouse Cedex 3, France b Dept. of Physiology, The Wenner-Gren Institute, The Arrhenius Laboratories F3, Stockholm University, SE-10691 Stockholm, Sweden article info abstract Article history: In mammalian cells, elongases and desaturases play critical roles in regulating the length and degree of Received 19 November 2009 unsaturation of fatty acids and thereby their functions and metabolic fates. In the past decade, a great Received in revised form 9 December 2009 deal has been learnt about these enzymes and the first part of this review summarizes our current knowl- Accepted 10 December 2009 edge concerning these enzymes. More recently, several transgenic mouse models lacking either an elong- ase (Elovl3À/À, Elovl4À/À, Elovl5À/À, Elovl6À/À) or a desaturase (Scd-1À/À, Scd-2À/À, Fads2À/À) have been developed and the second part of this review focuses on the insights gained from studies with these mice, Keywords: as well as from investigations on cell cultures. Elongase Ó 2009 Elsevier Ltd. All rights reserved. Desaturase VLCFA PUFA Knockout mice Contents 1. Introduction ......................................................................................................... 187 2. Mammalian elongases and desaturases . ...................................................................... 188 2.1. Elongases . ............................................................................................... 188 2.2. -

Prolyl-4-Hydroxylase Domain 3 (Phd3) Is a Critical Terminator for Cell Survival of Macrophages Under Stress Conditions
PROLYL-4-HYDROXYLASE DOMAIN 3 (PHD3) IS A CRITICAL TERMINATOR FOR CELL SURVIVAL OF MACROPHAGES UNDER STRESS CONDITIONS DOCTORAL THESIS In partial fulfillment of the requirements for the degree "Doctor of Philosophy (Ph.D.)" in the Molecular Medicine Study Program at the Georg-August University Göttingen Submitted by Lija Swain Born in Bhubaneswar, Odisha, India Göttingen, May 2014 i Members of the thesis committee Supervisor Prof. Dr. Dörthe Katschinski Göttingen University Medical Center Institute of Cardiovascular Physiology Humboldtallee 23, 37073 Göttingen, Germany Tel: +49 (0) 551 39-58 96 E-mail: [email protected] Co-supervisor Prof. Dr. Blanche Schwappach Göttingen University Medical Center Institute of Biochemistry I Humboldtallee 23, 37073 Göttingen, Germany Tel: +49 (0)551-39 5962 E-mail: [email protected] Co-supervisor Dr. Viacheslav Nikolaev Göttingen University Medical Center Department of Cardiology and Pneumology Robert-Koch-Str. 40, 37075 Göttingen, Germany Tel: +49 (0)551-39 10965 E-mail: [email protected] Date of disputation 7th July 2014 ii Dedicated to my Father iii Affidavit I hereby declare that my doctoral thesis entitled “Prolyl-4-hydroxylase domain 3 (PHD3) is a critical terminator for cell survival of macrophages under stress conditions” has been written independently with no other sources and aids than quoted. Lija Swain Göttingen, May 2014 iv Table of Contents Table of Contents Acknowledgement viii List of figures ix Abbreviations x Summary xiii 1. Introduction 1 1.1 Hypoxia 1 1.2 Hypoxia-inducible factors 1 1.2.1 HIF-1α 2 1.2.2 HIF-2α 3 1.2.3 HIF-3α 4 1.2.4 HIF-1β 4 1.3 Regulation of HIFα 5 1.4 Prolyl-4-hydroxylase Domain Enzyme (PHD) 8 1.4.1 PHD1 10 1.4.2 PHD2 10 1.4.3 PHD3 10 1.5 PHDs targets other than HIFs 13 1.6 Hypoxia and myeloid cells 15 1.7.1 Hypoxia and macrophages 15 1.8 Angiopoietin-like protein 2 (Angptl2) 17 1.9 Angptl2 and inflammation 18 1.10 Aim of the study 20 2. -

(FADS) Genetic Variants and Dietary Polyunsaturated Fatty Acid Intake: Associations with Negative Affect
Fatty Acid Desaturase (FADS) Genetic Variants and Dietary Polyunsaturated Fatty Acid Intake: Associations with Negative Affect Dissertation Presented in Partial Fulfillment of the Requirements for the Degree Doctor of Philosophy in the Graduate School of The Ohio State University By Liisa Hantsoo, M.A. Graduate Program in Psychology The Ohio State University 2012 Dissertation Committee: Janice K. Kiecolt-Glaser, Ph.D., Advisor Charles Emery, Ph.D. Ruchika Prakash, Ph.D. Copyrighted by Liisa Hantsoo 2012 Abstract Background: Depressive symptomatology has been associated with polyunsaturated fatty acid (PUFA) levels in diet and tissue. However, results have been mixed, and studies have failed to account for genetic factors that may influence such associations. Tissue PUFA levels are strongly influenced by elongase and desaturase activity, which are governed by the fatty acid desaturase (FADS) genes. Inefficient desaturase activity has been associated with depression. Further, FADS genotypes have been associated with neuropsychological phenotypes such as postpartum depression. Given these associations, the study of relationships among dietary PUFA intake, FADS genotype, and mood symptoms is warranted. Aims: The present study investigated associations among dietary intake of PUFAs, the rs174575 polymorphism of the fatty acid desaturase (FADS2) gene, and psychological outcomes. The primary aim was to determine if FADS2 genotype interacts with omega-3 (n-3) and omega-6 (n-6) PUFA dietary intake to influence associations with depressive symptoms, anxiety symptoms, anger, hostility, neuroticism, or optimism. Methods: A sample of 172 female undergraduate students provided genetic material from buccal cells, and completed a food frequency questionnaire and self-report measures including the Center for Epidemiological Studies Depression Scale, PROMIS Anxiety and Anger Scales, Cook Medley Hostility Scale, NEO Neuroticism Scale, and LOT-R Optimism Scale. -

Vitro Generated to Ex Vivo CD8 T Cells Comparative and Functional
Downloaded from http://www.jimmunol.org/ by guest on September 27, 2021 is online at: average * The Journal of Immunology , 21 of which you can access for free at: 2012; 189:3411-3420; Prepublished online 27 from submission to initial decision 4 weeks from acceptance to publication Comparative and Functional Evaluation of In Vitro Generated to Ex Vivo CD8 T Cells Dzana D. Dervovic, Maria Ciofani, Korosh Kianizad and Juan Carlos Zúñiga-Pflücker J Immunol August 2012; doi: 10.4049/jimmunol.1200979 http://www.jimmunol.org/content/189/7/3411 cites 61 articles Submit online. Every submission reviewed by practicing scientists ? is published twice each month by Receive free email-alerts when new articles cite this article. Sign up at: http://jimmunol.org/alerts http://jimmunol.org/subscription Submit copyright permission requests at: http://www.aai.org/About/Publications/JI/copyright.html http://www.jimmunol.org/content/suppl/2012/08/28/jimmunol.120097 9.DC1 This article http://www.jimmunol.org/content/189/7/3411.full#ref-list-1 Information about subscribing to The JI No Triage! Fast Publication! Rapid Reviews! 30 days* Why • • • Material References Permissions Email Alerts Subscription Supplementary The Journal of Immunology The American Association of Immunologists, Inc., 1451 Rockville Pike, Suite 650, Rockville, MD 20852 Copyright © 2012 by The American Association of Immunologists, Inc. All rights reserved. Print ISSN: 0022-1767 Online ISSN: 1550-6606. This information is current as of September 27, 2021. The Journal of Immunology Comparative and Functional Evaluation of In Vitro Generated to Ex Vivo CD8 T Cells Dzˇana D. Dervovic´,*,† Maria Ciofani,*,†,1 Korosh Kianizad,*,† and Juan Carlos Zu´n˜iga-Pflu¨cker*,† The generation of the cytotoxic CD8 T cell response is dependent on the functional outcomes imposed by the intrathymic constraints of differentiation and self-tolerance. -

Molecular and Functional Characterization of a Fads2 Orthologue in the Amazonian Teleost, Arapaima Gigas
Comparative Biochemistry and Physiology, Part B 203 (2017) 84–91 Contents lists available at ScienceDirect Comparative Biochemistry and Physiology, Part B journal homepage: www.elsevier.com/locate/cbpb Molecular and functional characterization of a fads2 orthologue in the Amazonian teleost, Arapaima gigas Mónica Lopes-Marques a,b, Rodrigo Ozório a,b, Ricardo Amaral c, Douglas R. Tocher d, Óscar Monroig d,⁎,1,L.FilipeC.Castroa,e,⁎,1 a CIIMAR – Interdisciplinary Centre of Marine and Environmental Research, U. Porto – University of Porto, Porto, Portugal b ICBAS - Institute of Biomedical Sciences Abel Salazar, U. Porto - University of Porto, Portugal c Universidade Federal do Acre, Brazil d Institute of Aquaculture, Faculty of Natural Sciences, University of Stirling, Stirling FK9 4LA, Scotland, UK e Department of Biology, Faculty of Sciences, U. Porto - University of Porto, Portugal article info abstract Article history: The Brazilian teleost Arapaima gigas is an iconic species of the Amazon. In recent years a significant effort has been Received 8 June 2016 put into the farming of arapaima to mitigate overfishing threats. However, little is known regarding the nutrition- Received in revised form 8 September 2016 al requirements of A. gigas in particular those for essential fatty acids including the long-chain polyunsaturated Accepted 27 September 2016 fatty acids (LC-PUFA) eicosapentaenoic acid (EPA) and docosahexaenoic acid (DHA). The ability to biosynthesize Available online 29 September 2016 LC-PUFA is dependent upon the gene repertoire of fatty acyl desaturases (Fads) and elongases (Elovl), as well as their fatty acid specificities. In the present study we characterized both molecularly and functionally an Keywords: Arapaima gigas orthologue of the desaturase fatty acid desaturase 2 (fads2)fromA. -
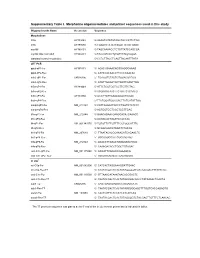
Supplementary Table I. Morpholino Oligonucleotides and Primer Sequences Used in This Study
Supplementary Table I. Morpholino oligonucleotides and primer sequences used in this study Oligonucleotide Name Accession Sequence Morpholinos tlr5a AY389449 5'-AAAGTGTATGTAGCTGCCATTCTGG tlr5b AY389450 5'-TGAATGTATATCCCATTCTGTGAGC myd88 AY388401 5'-TAGCAAAACCTCTGTTATCCAGCGA myd88 5bp mismatch AY388401 5'-TAcCAtAACCTgTGTTATCgAGgGA standard control morpholino 5'-CCTCTTACCTCAGTTACAATTTATA qRT-PCR ppial-qP1-Fw AY391451 5’- ACACTGAAACACGGAGGCAAAG ppial-qP2-Rev 5’- CATCCACAACCTTCCCGAACAC irak3-qP1-Fw CK026195 5’- TGAGGTCTACTGTGGACGATGG irak3-qP2-Rev 5’- ATGTTAGGATGCTGGTTGAGTTGG tlr5a-qP1-Fw AY389449 5’-ATTCTGGTGGTGCTTGTTGTAG tlr5a-qP2-Rev 5’-ACGAGGTAACTTCTGTTCTCAATG tlr5b-qP3-Fw AY389450 5’-GCGTTGTTGAAGAGGCTGGAC tlr5b-qP4-Rev 5’-TTCTGGATGGCCACTTCTCATATTGG mmp9-qP3-Fw NM_213123 5’-CATTAAAGATGCCCTGATGTATCCC mmp9-qP4-Rev 5’-AGTGGTGGTCCGTGGTTGAG il1b-qP1-Fw NM_212844 5’-GAACAGAATGAAGCACATCAAACC il1b-qP2-Rev 5’-ACGGCACTGAATCCACCAC il8-qP1-Fw XM_001342570 5’-TGTGTTATTGTTTTCCTGGCATTTC il8-qP2-Rev 5’-GCGACAGCGTGGATCTACAG ifn1-qP3-Fw NM_207640 5’- TTAATACACGCAAAGATGAGAACTC ifn1-qP4-Rev 5’- GCCAAGCCATTCGCAAGTAG tnfa-qP5-Fw NM_212829 5’- AGACCTTAGACTGGAGAGATGAC tnfa-qP6-Rev 5’- CAAAGACACCTGGCTGTAGAC cxcl-C1c-qP1-Fw NM_001115060 5’- GGCATTCACACCCAAAGCG cxcl-C1c-qP2_Rev 5’- GCGAGCACGATTCACGAGAG * In situ ccl-C5a-Fw NM_001082906 5’- CATCACTAGGAAAGGATTGAAC ccl-C5a-Rev-T7 5’- TAATACGACTCACTATAGGGGATGTCAAAGACTTTATTCAC cxcl-C1c-Fw NM_001115060 5’- GTTAAACATAAATAACACCGACTC cxcl-C1c-Rev-T7 5’- TAATACGACTCACTATAGGGACACCCTATAAAACTGAGTA irak3-Fw CK026195 5’- CAGTGAGAGAGGCATGAAACATC